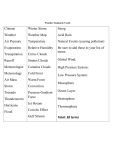
Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
10 ENVIRONMENTAL TECHNOLOGIES 10.1 Atmospheric chemistry 10.1.1 Introduction In recent years, growing concern about environmental issues has focused attention on the quality of atmospheric air, both in the troposphere, in other words from the ground up to an altitude of about 10 km, and in the stratosphere, due to the well-known and worrying phenomenon of the decrease in the concentration of ozone and the consequent thinning of the layer protecting the Earth from ultraviolet radiation. This interest has resulted in the creation of specific environmental regulations, which made their first appearance in the middle of the Twentieth century. In Europe, this legislation sets out limiting values and objectives to be reached by a certain date in the form of Directives (among the Directives currently in force it is worth recalling 96/62/EC, 99/30/EC, 2000/69/EC and 2002/3/EC). Their adoption by each member state means that these Directives become state law. Additionally, on an international level, commissions have been established to deal with specific problems and to draft protocols (the most important include the Geneva Protocol of 1979, the Montreal Protocol of 1987 and the Kyoto Protocol of 1997, with their subsequent amendments) subject to ratification by individual countries. An attempt has thus been made to take account of the impossibility of restricting to a single state or continent problems which inevitably affect neighbouring states (so-called transboundary pollution) or which, in the case of extremely complex phenomena such as climate change or the depletion of stratospheric ozone, affect the entire planet on a global scale. The latter two phenomena have been and remain (especially the first) an object of study by the scientific community and of interest to the authorities charged with managing the environment. The depletion of stratospheric ozone will VOLUME III / NEW DEVELOPMENTS: ENERGY, TRANSPORT, SUSTAINABILITY be dealt with below, as part of a brief overview of the complex interactions between the atmospheric ‘vessel’ and the compounds of natural and man-made origin emitted into it, thus creating a complex system. This system can only be understood by taking into account both the chemistry of the aforementioned compounds and the meteo-climatic factors which can alter their concentrations, for example by preventing their mixing and consequently their dispersion (episodes of thermal inversion are in fact closely correlated with cases of extreme pollution). There are four basic processes underlying pollution phenomena: emission, transformation, diffusion and transportation, and deposition. The first two processes essentially concern those pollutants described as ‘primary’, in other words those emitted directly as such, which may then undergo transformations and give rise to ‘secondary’ pollutants (such as nitrogen dioxide and ozone). These two classes of pollutants share their subsequent ‘itinerary’, which depends on the dynamic behaviour of the lower layers of the atmosphere, as far as transportation and diffusion mechanisms are concerned, and is heavily conditioned by this and by other meteo-climatic parameters (such as relative humidity), as far as sink processes through dry and wet deposition are concerned. This chapter focuses on the chemistry of the compounds of greatest environmental interest (nitrogen oxides, sulphur dioxide, non-methane organic compounds, ozone). Specifically, the reactions characterizing these in both the gas phase (homogeneous reactions) and in the aqueous phase (heterogeneous reactions) will be examined: the presence of water in the atmosphere, in different states of aggregation, is important for general considerations on the behaviour of these species after their release into the atmosphere. 915 ENVIRONMENTAL TECHNOLOGIES An in-depth understanding of all the processes governing pollution phenomena is essential both for the ability to predict how these will evolve in the short and long term, and to identify and implement appropriate control strategies. (or nitrous oxide), nitrogen trioxide, tetroxide and pentoxide. Nitrogen monoxide is a colourless and odourless primary pollutant which forms from combustion at about 1,200°C, whilst nitrogen dioxide is a reddish pollutant with a strong and pungent smell. Nitrogen dioxide is a secondary pollutant since it does not form directly from combustion (except in a percentage of 4-5%). The sequence of reactions leading to the formation of nitrogen oxides is described below. Combustion produces NO (primary pollutant) through the reaction: 10.1.2 Atmospheric chemistry in the gas phase The apparent stability of the atmosphere derives from the fact that it is in a stationary state; this situation is due to the relative constancy of inputs and outputs which on average balance each other out. Since most sources and sinks of the gases in the atmosphere are the result of chemical reactions, an understanding of reaction rates in the gas phase is extremely important. However, the study of chemical kinetics concentrates not only on reaction rates but also on determining the precise reaction mechanism. This paragraph provides a brief survey of the reactions of the main pollutants in the gas phase. Specifically, Fig. 1 shows the reactions induced by the chemical species most heavily involved in the formation of acidic compounds in the atmosphere. [1] 䉴 which in turn can produce NO2 (secondary pollutant) via the (non-photochemical) thermal oxidation reaction: [2] 2NO ⫹O2⫺ 2NO2 䉴 However, reaction [2] is of minor importance since it is too slow at the concentrations of NO normally present in the atmosphere. The reaction kinetics is of the second order with respect to NO and of the first order with respect to O2; therefore, the kinetic equation is R⫽k [O2] [NO]2, where R is the reaction rate and k is the rate constant (equal to 2.0 ⭈10⫺38 cm6 molecules⫺2 s⫺1). Nitrogen dioxide is not produced only as shown above; its formation is also triggered by ultraviolet radiation and tropospheric ozone. Nitrogen oxides The term nitrogen oxides is generally used to describe a mixture of nitrogen monoxide (95% of the total), nitrogen dioxide and traces of dinitrogen oxide Fig. 1. Some reactions which occur in the atmosphere induced by NOx, SO2 and NH3 (the species most involved in the formation of acid compounds in the atmosphere and in their subsequent salification). N2⫹O2⫺ 2NO O3 O3 NO aerosol OH HNO3 NO2 hn nitrate aerosol NH3 emission dry deposition OH wet deposition NH3 SO2 H2SO4 sulphate aerosol H2O2 emission dry deposition wet deposition NHO3 ammonium aerosol NH3 H2SO4 emission 916 dry deposition wet deposition ENCYCLOPAEDIA OF HYDROCARBONS ATMOSPHERIC CHEMISTRY The photolysis of NO2 produces ozone through the reactions: [3] [4] NO2 ⫹hv (l ⭐430 nm)⫺ NO ⫹O(3P) O(3P) ⫹O2⫺ O3 (k ⫽6.0 ⭈10⫺34 (T/300) ⫺2,3 cm6 molecules⫺1s⫺1) 䉴 䉴 where hv is the light radiation and l is the wave length. The reaction between ozone and nitrogen monoxide then produces nitrogen dioxide: O3 ⫹NO⫺ NO2 ⫹O2 (k ⫽1.8 ⭈10⫺14 cm3 molecules⫺1s⫺1) There is not therefore a net loss of ozone, since the titration reaction causes it to be destroyed whilst the NO2 photolysis reaction simultaneously causes reemission; it can thus be deduced that the three species are in fact involved in a photostationary equilibrium. The rate constant in decomposition processes by photolysis of a trace species present in the atmosphere, whose dimensions are the inverse of time (s⫺1), can be generically defined as: [5] [6] 䉴 冮 J ⫽ s(l)f(l)j(l)dl where s(l) is the absorption cross-section expressed in cm2 molecules⫺1, which is characteristic of each chemical species, f(l) is the quantum yield of the photolysis reaction, and finally j(l) is the actinic flux (number of photons⭈cm⫺2⭈s⫺1, generally used instead of I⬘i ⫽I⬘r⫹I⬘s⫹I⬘d); in other words it is the product of the components of the solar radiation incident (I⬘i ) directly on the air masses, consisting of reflected radiation (I⬘r ), scattered radiation (I⬘s ), and finally that which reaches the Earth’s surface directly (I⬘d ). Assuming the absence of organic compounds in the atmosphere, the relationship between the concentrations of O3, NO and NO2 at a generic time t in a mass of air is a constant given by the ratio of JNO2 (the rate constant of NO2 photolysis) to kNO (the rate constant of reaction [5] between NO and ozone): [7] JNO2[NO2] [O3]⫽ 11112 kNO[NO] The constant JNO2 varies depending on the angle of the luminous solar radiation, and so the relationship between the concentrations changes over the course of a day. The average value of JNO2 is 0.75⭈10⫺2 s⫺1 or 0.533 min⫺1. The presence of RO⭈2 and HO⭈2 radicals, which form mainly during the hot season, promotes reactions with nitrogen monoxide, in other words: [8] RO⭈⫹NO⫺ NO2 ⫹RO 2 (if R ⫽CH3CH2CH2, k ⫽7.6 ⭈10⫺12 cm3 molecules⫺1s⫺1) 䉴 VOLUME III / NEW DEVELOPMENTS: ENERGY, TRANSPORT, SUSTAINABILITY [9] HO2⭈⫹NO⫺ HO ⫹NO2 (k ⫽8.3 ⭈10⫺12 cm3 molecules⫺1s⫺1) 䉴 As a result, under these conditions, reaction [5] is hindered and O3, being unable to react with NO, accumulates in the lower levels of the atmosphere. In the presence of NO2, high concentrations of RO⭈2 and HO⭈2 free radicals give rise to alkyl nitrate hydroperoxides, PeroxyAcetyl Nitrates (PAN) and PeroxyBenzoyl Nitrates (PBN). PAN is a gas which tends to accumulate persistently in the coldest parts of the troposphere; it then spreads to warmer areas, leading to the formation of free radicals and NO2 since the dissociation constant of PAN depends strongly on temperature. World Health Organization (WHO) guidelines state that the annual averages of NOx in European cities are around 40 mg/m3 and range from 20 to 90 mg/m3 in industrialized countries; the base level is between 1 and 9 mg/m3. Nitrogen dioxide is more toxic than nitrogen monoxide. For this reason, NO2 rather than NO is monitored by law; values of 13 ppm cause irritation to the mucous membranes of the nose and eyes; exposure to concentrations of 560 mg/m3 for 30 minutes causes pulmonary problems. The WHO recommends that hourly mean concentrations of 200 mg/m3 and an average annual limit value of 40 mg/m3 should not be exceeded. As already seen, NO2 is a pollutant which is mainly generated indirectly from the NO emitted by the combustion of fuels used in road transportation and is thus described as a ‘mobile source pollutant’. It is found mainly in urban areas with a high traffic density, coinciding with the opening and closing times of workplaces and schools, and is also present in large car parks. Another source of this substance is combustion processes in civil and industrial plants. As well as being harmful to human health, nitrogen dioxide acidifies rain, degrades man-made objects, corrodes metals and damages vegetation. During the day, NO2 is oxidized to nitric acid by reaction with the ⭈OH radical: [10] NO2 ⫹⭈OH ⫹M⫺ HNO3 ⫹M (k ⫽1.1⭈10⫺11 cm3 molecules⫺1s⫺1) 䉴 where M is a third ‘body’ or molecule (typically N2 or O2) that has the role of removing the excess energy through collision and so influences the kinetics of the reaction itself. This reaction is slow compared to the NO-NO2 exchange: the average life-time of NOx is typically 1-2 days in the low troposphere at middle latitudes. The nitric acid is then deposited on the Earth’s surface through various mechanisms. 917 ENVIRONMENTAL TECHNOLOGIES The NO3 radical is particularly important in the chemistry of organic compounds during the night, when its average life-time increases since it does not undergo photodissociation (as occurs during the day) and is thus able to react with Volatile Organic Compounds (VOCs), oxidizing them rapidly. Its action is similar to that of ⭈OH during the daytime. The reactions which take place can be grouped as follows: • formation reactions: [11] [12] [13] • 䉴 䉴 NO2 ⫹NO3 ⫹M⫺ ⫺N2O5 ⫹M 䉳 䉴 For this reaction, the equilibrium constant reported in the literature varies by a factor of 1.9 at ambient temperature (from 1.8⭈10⫺11 to 3.44⭈10⫺11 cm3 molecules⫺1s⫺1). The N2O5 thus obtained reacts with water to give nitric acid: 䉴 NO3 ⫹hv (l ⭐640 nm)⫺ NO2 ⫹O(3P) NO3 ⫹hv (585 ⭐ l ⭐640 nm)⫺ NO ⫹O2 䉴 䉴 reactions with organic compounds [16] NO3 ⫹RH⫺ HNO3 ⫹R [17] NO3 ⫹RCHO⫺ HNO3 ⫹RCO [18] NO3 ⫹C⫽C⫺ ⫺C(ONO2)⫺C⫺ 䉴 䉴 䉴 Addition reactions to alkenes (especially biogenic isoprene and monoterpenes) are much faster than those which occur with alkanes. The reaction of the nitrate with olefins also forms peroxide radicals (HO⭈2 ⫹RO⭈) 2 and it has been shown (Salisbury et al., 2001) that NO3 may be as important in their formation as O3. The formation of nitric acid due to reaction with hydrocarbons accounts for about 15% of the nitric acid in the atmosphere. Considering the average concentrations of these organic compounds in the atmosphere (in typically polluted urban atmospheres alkanes are present in concentrations of about 100 ppb whilst formaldehyde and acetaldehyde are present respectively in concentrations of 20 ppb and 10 ppb) and the rate constants, and assuming that the concentration of NO3 is 100 ppt, the net overall rate of formation is about 0.3 ppb h⫺1. Making similar assumptions for an average concentration of NO2 of 50 ppb and ⭈OH concentrations of 1⭈106 cm⫺3 (typical of a moderately polluted atmosphere) the rate of formation is 2 ppb h⫺1. The nitrate is thus a sink for NOx during the night, and may in turn be removed by various mechanisms involving other sinks such as reactions with organic compounds or deposition on aerosols or on the ground (direct sink). It has in fact been shown that the average life-time of NO3 depends on the relative humidity decreasing rapidly to less than 10 minutes when 918 [19] [20] photodissociation reactions [14] [15] • NO ⫹O3⫺ NO2 ⫹O2 (k ⫽1.8 ⭈10⫺14 cm3 molecules⫺1s⫺1) NO2 ⫹O3⫺ NO3 ⫹O2 (k ⫽3.2 ⭈10⫺17 cm3 molecules⫺1s⫺1) NO2 ⫹⭈OH⫺ HNO3 (k ⫽1.1⭈10⫺11 cm3 molecules⫺1s⫺1) relative humidity is 50% (Platt et al., 1984). This is probably due to interaction with the water found on the surfaces of the particles present in the environment. In part, this may also be explained by the reaction of nitrogen pentoxide (N2O5) which represents an indirect sink for NO3 and which may react in the presence of water, shifting the equilibrium: N2O5 ⫹H2O⫺ 2 HNO3 (k ⭐1.3 ⭈10⫺21 cm3 molecules⫺1s⫺1) 䉴 Homogeneous hydrolysis thus represents a sink for NO3 and at the same time a source of nitric acid: although the rate constant seems fairly low, this reaction contributes significantly to the formation of nitric acid in the homogeneous phase (0.3 ppb h⫺1 at 50% of relative humidity) as well as in the heterogeneous phase, and in this case is significantly faster. Essentially, the main formation paths for nitric acid are the reaction between NO2 and ⭈OH, the reaction between nitrate and organic compounds and the hydrolysis of N2O5. Nitric acid has a fairly long life-time, and may thus be the terminal of various chain reactions in the troposphere. It absorbs weakly in the actinic region and thus does not undergo photolysis, however it may undergo deposition (dry and wet) and react with the ⭈OH radical and (albeit slowly) with ammonia: [21] [22] HNO3 ⫹⭈OH⫺ H2O ⫹NO3 HNO3 ⫹NH3⫺ NH4NO3 䉴 䉴 Ammonium nitrate is thus in equilibrium with the two species in the gas phase, and this equilibrium exists for both ammonium in the solid phase (relative humidity⬍62%) and in solution (relative humidity⬎62%). Sulphur dioxide (SO2) Sulphur dioxide (SO2) is a colourless gas with an acrid, pungent smell. Emissions of this gas are mainly due to the use of solid and liquid fuels and are directly correlated with their sulphur content: [23] S ⫹O2⫺ SO2 䉴 This is, therefore, a typical pollutant of industrial and urban areas, in the latter particularly during the winter (due to domestic heating). Natural concentrations of SO2 are less than 5 mg/m3; annual ENCYCLOPAEDIA OF HYDROCARBONS ATMOSPHERIC CHEMISTRY averages are lower than 50 mg/m3; and daily averages rarely exceed 125 mg/m3. Sulphur dioxide is used as a ‘tracer’, in other words a global indicator of atmospheric pollution, due to its chemical stability in the atmosphere. Most SO2 undergoes chemical transformations before it reaches the ground; it is oxidized to SO3, followed by hydrolysis to H2SO4, particles of which absorb further SO2, NH3 and traces of metals to form a particulate aerosol which, depending on weather conditions, may be transported for hundreds of kilometres and reach the ground in the form of acid rain. The oxidation reaction of SO2: [24] 2SO2 ⫹O2⫺ 2SO3 䉴 has such a low rate in the absence of catalysts that it can be completely disregarded as a source of SO3; the same can be said for photooxidation as a reaction mechanism, since if every SO*2 molecule in an excited state was oxidized by reaction with O2 or other species, the average life-time of SO2 in the low troposphere would be 52 minutes, which is, in fact, not the case. In any case, the only rapid gas phase process which is efficient enough to account for most of the sulphuric acid present in the aerosols formed by gas phase processes is the reaction of SO2 with the ⭈OH radical: [25] SO2 ⫹⭈OH ⫹M⫺ HOSO⭈⫹M 2 (k ⫽1.1⭈10⫺12 cm3 molecules⫺1s⫺1) 䉴 It is known that a significant fraction of the HOSO⭈2 radical is eventually transformed into sulphuric acid, but the rate of the reaction and its intermediate products are not well known; as such, the reaction is generically described as: [26] HOSO⭈⫺ ⫺ ⫺ H2SO4 2 䉴 䉴 䉴 In any case, a mechanism by which ⭈OH is regenerated has been suggested: [27] [28] HOSO2 ⫹O2⫺ HO2⭈⫹SO3 HO⭈⫹NO⫺ NO2 ⫹OH 2 䉴 䉴 This mechanism was proposed by Calvert and Stockwell (1984), on the basis of the experimental evidence that in a photooxidant mixture of HNO2, NO, NO2 and CO, even the addition of substantial amounts of SO2 does not influence the concentration of ⭈OH. The oxidation of SO2 by this mechanism averaged out over 24 hours is 16.4%; during the winter the rate is lower due to the lower concentration of ⭈OH. Other oxidation reactions induced by other oxidizing species, such as O (3P), HO⭈2 and CH3O⭈, 2 are characterized by lower rate constants than the reaction with ⭈OH, which remains the main oxidation reaction induced by SO2 (the rate constants are k⫽5.7⭈10⫺14, k⬍1⭈10⫺18, and k⬍1⭈10⫺18 respectively). VOLUME III / NEW DEVELOPMENTS: ENERGY, TRANSPORT, SUSTAINABILITY Levels of SO2 are generally far higher than those of SO3 since the latter, in contact with water vapour, leads to the following reaction, also encouraged by the presence of particulate matter and solar radiation: [29] SO3⫹H2O⫺ H2SO4 (k ⫽9.1⭈10⫺13 cm3 molecules⫺1s⫺1) 䉴 Naturally, other fundamentally important oxidation reactions of SO2 to sulphuric acid are those which take place in the aqueous phase inside the water droplets present in the atmosphere; these will be dealt with in the paragraph on solution equilibria. Non-methane volatile organic compounds Methane is the most abundant hydrocarbon present in the Earth’s atmosphere and the most stable with respect to attack by ⭈OH. This means that it can be transported far from its source before it is destroyed. By marked contrast, terpenes are extremely reactive and as a result have short life-times. A broad variety of hydrocarbons, the so-called Biogenic Non-Methane Hydrocarbons (BNHC), are emitted by natural sources: these are unsaturated organic compounds emitted mainly by plants, such as isoprene and some monoterpenes like a-pinene, b-pinene, d-limonene, etc. In towns the air may contain variable concentrations (even above 1.0-2.0 mg/m3) of hydrocarbons other than methane, whilst methane itself may exceed 1.5 mg/m3. The most important reactions involving these species (RH) are those which lead to the formation of ozone: [30] RH⫹⭈OH ⫹O2⫺ RO⭈(⫹H 2O) 2 [31] RO ⫹NO2 RO⭈⫹NO⫺ 2 [32] NO2 ⫹O2 ⫹hv⫺ O3 ⫹NO 䉴 䉴 䉴 As such, the amount of ozone produced per mole of hydrocarbon depends on its concentration and its reactivity with ⭈OH. The relative reactivity of species j in a mixture of i hydrocarbons (HC) can thus be expressed as: kj [HC]j [33] Rj ⫽ 111133 冱ki[HC]i i Taking account of the typical abundances of these compounds and their reactivity, it is possible to calculate the rates of degradation. For example, Table 1 shows the removal rates of some of the most abundant hydrocarbons in the atmosphere. In terms of the rate of degradation, alkenes have the highest potential for forming ozone, followed by aromatic compounds, whilst alkanes have the lowest formation potential. 919 ENVIRONMENTAL TECHNOLOGIES Table 1. Kinetic data on the removal of hydrocarbons present in the atmosphere by ⭈OH radicals k at 298K (10⫺12 cm3, molec.⫺1s⫺1) Concentration (1010 molec. cm⫺3) Removal rate (10⫺2s⫺1) Methane 0.0077 5,748.0 44.3 Toluene 6.4 12.1 77.4 Ethylene 8.8 26.8 235.8 Acetylene 0.9 16.1 14.5 Benzene 1.0 5.3 5.3 COMPOUND The oxidation of hydrocarbons is strictly correlated with the photostationary state NOx⫺O3 described by the Leighton relationship: the oxidation of VOCs (Fig. 2) by ⭈OH produces the radical RO⭈2 which converts NO to NO2 through the reaction already described for NOx without involving the simultaneous removal of O3. As a result, the concentration of ozone increases, forming other ⭈OH radicals, thus increasing the rate at which VOCs are oxidized. The maximum production of O3 requires an appropriate ratio of NOx to VOC concentrations: a lack of NOx results in an insufficient production of ⭈OH to induce the oxidation of VOCs, whilst a lack of VOCs makes it impossible to reach the concentrations of RO2⭈ needed to alter the photostationary state significantly. The non-linearity of ozone formation has been demonstrated experimentally by Kelly and Gunst (1990). Surveying the main reactions of the various classes of organic compounds, it can be seen that the main reactions are those with ⭈OH and NO3, whose constants for the various hydrocarbons are reported in Table 2. The chemistry of the higher term alkanes follows the same mechanism as methane: the attack by ⭈OH takes place preferentially to form the most stable alkyl radical, therefore tertiary and secondary hydrogens are those which react most easily with ⭈OH. The chemistry of anthropogenic alkenes follows a similar mechanism to that of biogenic isoprene: the initial reaction is the addition of ⭈OH followed by the addition of O2 to a hydroxy-substituted alkylperoxy radical, which in turn reacts with either NO or HO⭈2 depending on whether the concentrations of NO are high or low. The photochemical oxidation of carbonyl compounds leads to the production of peroxyacetyl radicals and the following reactions give rise to the formation of PAN (CH3C(O)O2NO2): [34] CH3CHO ⫹⭈OH (⫹O2)⫺ CH3C(O)O2 ⫹H2O (k ⫽1.6 ⭈10⫺11 cm3 molecules⫺1s⫺1) [35] CH3C(O)O2 ⫹NO2 ⫹M⫺ CH3C(O)O2NO2 ⫹M (k ⫽3.6 ⭈10⫺12 cm3 molecules⫺1s⫺1) 䉴 䉴 VOC ⫽RCH3 ⴢOH, O2, M RO2ⴢ ⫽RCH2O2 NO via aldehydes NO2 ⫹ ROⴢ ⫽RCH2O hn NO O O2 O3 Fig. 2. Schematic of the oxidation of organic compounds in the troposphere. 920 O2 O2 Table 2. Rate constants for VOC oxidation reactions R⫹CHO CONCENTRATION kOH(1012cm3 kNO3(1016cm3 molec.⫺1s⫺1) molec.⫺1s⫺1) (ppb carbon) hn COMPOUND RCHO Isopentane 45.3 3.9 1.6 Toluene 33.8 5.96 0.3 Ethylene 21.4 8.52 2.1 Acetylene 12.9 0.9 ⭐0.2 Benzene 12.6 1.23 0.2 Isoprene – 101 5,900 a-pinene – 53.7 58,000 ENCYCLOPAEDIA OF HYDROCARBONS ATMOSPHERIC CHEMISTRY [36] CH3C(O)O2NO2 ⫹M⫺ CH3C(O)O2 ⫹NO2⫹M (k ⫽1.8 ⭈10⫺4 cm3 molecules⫺1s⫺1) 䉴 The formation of PAN is a process which ends the propagation of the reaction chain, and which competes with the reaction between the peroxy radical and NO: [37] CH3C(O)O2 ⫹NO⫺ CH3C(O)O ⫹NO2 (k ⬎1⭈10⫺11cm3 molecules⫺1s⫺1) [38] CH3C(O)O ⫹O2⫺ CH3O2⭈⫹CO2 (k ⫽2.0 ⭈10⫺12 cm3 molecules⫺1s⫺1) [39] CH3O2⭈⫹NO⫺ CH3O ⫹NO2 (k ⫽7.6 ⭈10⫺12 cm3 molecules⫺1s⫺1) [40] CH3O ⫹O2⫺ HCHO ⫹HO2⭈ (k ⫽1.9 ⭈10⫺15 cm3 molecules⫺1s⫺1) 䉴 䉴 䉴 䉴 The formation of PAN is encouraged by low temperatures and pressures. Thermal decomposition is the most important destruction path for PAN near the Earth’s surface, whilst at altitudes above 7 km it reacts with ⭈OH: [41] CH3C(O)O2NO2⫹⭈OH⫺ products (k ⫽1.1⭈10⫺13cm3 molecules⫺1s⫺1) 䉴 Therefore, if the PAN formed rises rapidly to height atmosphere, its life-time increases and it may represent a source of NOx through long-distance transportation mechanisms. Ozone and photochemical smog For a photochemical smog process to be triggered, sunlight, nitrogen oxides and volatile organic compounds must be present; additionally, the process is favoured by a high atmospheric temperature. Since nitrogen oxides and volatile organic compounds are among the main components of emissions in urban areas, towns located in geographical areas characterized by intense solar radiation and high temperatures (such as those in the Mediterranean) are ideal candidates for episodes of intense photochemical pollution. The knowledge necessary for understanding secondary pollution events thus concerns the chemical and chemico-physical transformation processes undergone by pollutants, dynamic processes in the lower atmosphere (atmospheric stability, direction and intensity of the wind) and the intensity of solar radiation. Ozone is a photochemical oxidant similar to PAN, nitrogen dioxide and hydrogen peroxide which are essentially secondary pollutants formed in the troposphere by chemical reactions starting from primary pollutants (basically VOCs and nitrogen oxides which are therefore also described as precursors) in the presence of solar radiation. It is naturally present in the troposphere at concentrations ranging from 20 to 80 ppb. VOLUME III / NEW DEVELOPMENTS: ENERGY, TRANSPORT, SUSTAINABILITY Ozone has a characteristic smell and may cause severe irritation to the respiratory system and the eyes at concentrations exceeding 200 ppb. It is also a cause of the oxidative degradation of some non-biological materials, especially elastomers, textile fibres and dyes. In fact, the presence of only six electrons in the valence shell of oxygen gives it electrophilic properties and therefore the tendency to remove electrons from other species or to share them. It is characterized by a redox potential of 2.07 V in an aqueous system. The formation reactions of ozone by nitrogen oxides and hydrocarbons have already been reported extensively in the chapter devoted to those pollutants. In the lower atmosphere, ozone forms from the reaction of atmospheric oxygen with the atomic oxygen produced by the photolysis of nitrogen dioxide; the ozone formed is in turn removed by nitrogen monoxide, with the new formation of NO2. In unpolluted atmospheres, where other chemical species are not present in appreciable quantities, this series of reactions forms a cycle (the photostationary ozone cycle) and there is no possibility for photochemical pollution. The fundamental step towards the atmospheric enrichment of ozone and other photooxidant species (in other words oxidizing chemical species formed by chemical reactions that occur only in the presence of light) is the formation of NO2 by alternative pathways which do not entail the removal of ozone. Identifying the formation pathways of NO2 thus represents the key to understanding photochemical oxidation processes. The main alternative pathway for the formation of NO2 is the oxidation of NO by peroxide radicals (RO⭈). 2 These free radicals originate from the degradation of volatile hydrocarbon molecules (RH) and their subsequent reaction with atmospheric oxygen. The attack on volatile hydrocarbons is due to the presence in the atmosphere of other free radicals, ⭈OH hydroxyl radicals: the processes which generate hydroxyl radicals are thus fundamental in triggering photochemical pollution processes. The production of ⭈OH radicals is essentially a photochemical process and the main precursors are nitrous acid, formaldehyde and ozone itself. Ozone is therefore not only the most quantitatively important product of photochemical pollution processes, but also part of the ‘fuel’ which activates the process. In fact, ozone, like nitrogen dioxide, undergoes photolysis and given the energy of the O2⫺O bond which is only 101 kJ mol⫺1 (in other words in the order of 1 eV per molecule) and since the energy E of a photon is linked to frequency n by the relationship E⫽hn, fairly low frequency radiation is needed (but in 921 ENVIRONMENTAL TECHNOLOGIES any case over 2.53⭈1014 Hz, in other words with a wavelength shorter than 1.185 nm) to cleave the bond; most of the spectrum of solar radiation thus has sufficient energy to cleave the O2⫺O bond: [42] O3 (1A1) ⫹hv (l⬍1,180 nm)⫺ O(3P) ⫹O2(3S g⫺) (J ⫽4.2 ⭈10⫺4 s⫺1) 䉴 At shorter wavelengths, the photon’s excess energy may be converted into the electronic excitation of the products: [43] O3 (1A1)⫹hv (l⬍310 nm)⫺ O(1D) ⫹O2(1Dg) (J ⫽2.9 ⭈10⫺5 s⫺1) 䉴 Electronically excited molecular oxygen (1Dg) may be another potential oxidizing agent in the troposphere, especially for unsaturated hydrocarbons: the rate constants for reactions with these compounds are in the order of 10⫺18 cm3 molecules⫺1s⫺1. Comparing this with the rate constants of the reactions of alkenes with ⭈OH and O3 (on the order of 10⫺12, as reported in Table 1, and 10⫺18 cm3 molecules⫺1s⫺1 respectively) leads to the conclusion that, to have a significant effect on tropospheric chemistry, the concentrations of O2 (1Dg) would need to be of the same order of magnitude as ozone or greater. In fact, concentrations of this species are around 10 ppt. Since the transition: [44] O O(1D) ⫹H2O⫺ 2⭈OH (k ⫽2.2 ⭈10⫺10 cm3 molecules⫺1s⫺1) 䉴 This reaction is extremely fast and occurs in competition with the deactivation of the O (1D) by air (indicated by M): [46] O(1D) ⫹M⫺ O (3P) ⫹M (k ⫽2.9 ⭈10⫺11 cm3 molecules⫺1s⫺1) 922 R(⭈OH, O3) ⫽4.65 ⭈105 molecules cm⫺3 s⫺1 HONO ⫹hv(l ⬍320 nm)⫺ ⭈OH ⫹NO (J ⫽1.8 ⭈10⫺3 s⫺1) 䉴 H2O2 ⫹hv(l ⬍360 nm)⫺ 2⭈OH (J ⫽6.9 ⭈10⫺6 s⫺1) In the case of formaldehyde, the predominant mechanism when 300⭐l ⭐320 nm is: [49] [50] 䉴 HCHO ⫹hv ⫺ H ⫹CHO (J ⫽1.7 ⭈10⫺5s⫺1) 䉴 Subsequent oxidation leads to the formation of the ⭈OH radical: [51] H ⫹O2 ⫹M⫺ HO⭈⫹M 2 (k ⫽7.0 ⭈10⫺13 cm3 molecules⫺1s⫺1) [52] CHO ⫹O2⫺ HO⭈⫹CO 2 (k ⫽6.0 ⭈10⫺12 cm3 molecules⫺1s⫺1) [53] HO⭈⫹NO⫺ ⭈OH ⫹NO2 2 (k ⫽8.0 ⭈10⫺12 cm3 molecules⫺1s⫺1) 䉴 䉴 䉴 At wavelengths above 340 nm there is a preferential dissociation into relatively stable products: [54] HCHO ⫹hv⫺ H2 ⫹CO (J ⫽4.3 ⭈10⫺5s⫺1) 䉴 Crutzen (1988) has estimated that on average 50-60% of the formaldehyde follows the mechanism above, 20-25% follows the mechanism leading to the formation of CHO and H and subsequent oxidation reactions, and 20-30% reacts directly with ⭈OH according to the reaction: [55] 䉴 In an atmosphere characterized by a relative humidity of 50% and a temperature of 298K, about 10% of the O (1D) produced reacts with water to form hydroxyl radicals. At middle latitudes, the main source of ⭈OH radicals is ozone, given the high concentrations of this species which is about 40 ppb or 9.84⭈1011 molecules cm⫺3. In fact, the rate of production of ⭈OH radicals from the photolysis of ozone at middle latitudes is: [47] [48] (1D)⫺䉴 O (3P) is impossible, as this is a spin-forbidden transition, O (1D) may undergo thermal degradation through collisional energy transfer or react with other species, like CH4 or H2O, extracting a proton from them and thus giving rise to ⭈OH radicals: [45] Other sources of ⭈OH radicals are the photolysis of nitrous acid, hydrogen peroxide and formaldehyde. Nitrous acid and formaldehyde are precursors of ⭈OH radicals, but in their turn have formation pathways which are essentially secondary, starting from species involved in photochemical processes (nitrogen dioxide for nitrous acid and hydrocarbons and radicals or ozone for formaldehyde). The formation reactions for ⭈OH radicals starting from nitrous acid and hydrogen peroxide are as follows: HCHO ⫹⭈OH⫺ HCO ⫹H2O (k ⫽1.0 ⭈10⫺11 cm3 molecules⫺1s⫺1) 䉴 The ⭈OH radical may also give rise to the oxidation of CO: [56] CO ⫹⭈OH⫺ CO2 ⫹H (k ⫽2.0 ⭈10⫺13 cm3 molecules⫺1s⫺1) [57] H ⫹O2 ⫹M⫺ HO⭈⫹M 2 (k ⫽7.0 ⭈10⫺13 cm3 molecules⫺1s⫺1) 䉴 䉴 In the presence of a source of NO, ⭈OH will therefore be obtained again directly via the oxidation reaction induced by the HO⭈2 radical, and indirectly via the photolysis of the ozone produced. The reactions involving formaldehyde also produce hydroperoxide radicals, which are among the radical ENCYCLOPAEDIA OF HYDROCARBONS ATMOSPHERIC CHEMISTRY species fundamental in photochemical smog processes alongside hydroxyl radicals and alkyl peroxide radicals (formed, as seen above, from organic compounds). There is an interconversion between hydroxyl radicals and hydroperoxide radicals, and the key reaction in this process is as follows: [58] HO⭈⫹NO⫺ ⭈OH ⫹NO2 2 䉴 Photochemical pollution is thus caused by a sequence of interdependent reactions (in some cases true chain reactions) which give rise to a process which feeds itself; this explains why acute episodes of photochemical smog often last for several consecutive days, increasing in intensity. Atmospheric secondary particulate matter The formation of aerosols in the atmosphere has a significant impact on visibility, the climate and the chemical processes which occur in the atmosphere; it is also of special interest since the finest fraction (with an aerodynamic diameter less than 2.5 mm) may penetrate the alveoli of the lungs and thus has a direct impact on human health. Aerosols in the troposphere may be emitted directly (primary particulate matter) or be formed from chemical processes (secondary particulate matter). The sources are both natural and man made, and the composition of this material therefore varies Fig. 3. Negative feedback cycle between UV radiation and particulate matter. considerably. The dimensions of the particles vary significantly for both primary and secondary particulate matter, with aerodynamic diameters ranging from 2 nm to over 10 mm. Secondary aerosols are generated by the gasparticle conversion which follows the formation, through oxidative processes, of products characterized by particularly low volatility or high solubility. Since these oxidative processes are often of a photochemical nature, the resulting aerosols may be counted among the secondary photochemical pollutants. Secondary aerosol may be generated either by condensation onto existing aerosol or by nucleation to form new particles or suspended droplets (Seinfeld, 1986; Clement and Ford, 1996). The most important molecule which may give rise to the nucleation of new particles is H2SO4. Fig. 3 summarizes the processes which occur in the presence of primary and secondary pollutants and UV radiation: the interactions between the latter and the particles have been the object of recent studies (Kikas et al., 2001). These have shown that the rate of photolysis of some pollutants (O3, NO2) depends on the presence of particles which absorb UV radiation (especially elementary carbon and organic aerosols; Jacobson, 1999) or scatter it (the optical properties of the particles vary with size and are strictly linked to their chemical composition). A study by Jacobson (1998) has shown that levels of UV radiation scattering gases scattering particulate matter absorbing gases absorbing particulate matter (NO3, HONO, PAN, RO2ⴢ, etc.) O3 NO2 HOⴢ SO2 HO⫺, aqueous oxidation H2SO4 NO HNO3 VOC HO O3, NO3, HOⴢ semi-volatile organics NH3 particulate sulfate VOLUME III / NEW DEVELOPMENTS: ENERGY, TRANSPORT, SUSTAINABILITY particulate nitrate secondary organic aerosol 923 ENVIRONMENTAL TECHNOLOGIES ozone in Los Angeles have fallen by 5-8% due to aerosols. Wendisch and colleagues (1996) have measured the vertical profile of particulate matter to calculate the vertical rate profile of the NO2 photolysis reaction. With regard to the composition of secondary particulate matter, studies have been undertaken exploiting the different optical properties (light scattering) at different relative humidities of the species of interest: H2SO4 and (NH4)2SO4. These studies have shown that ammonium sulphate is often observed in real samples, and there is therefore sufficient ammonia in ambient air to completely neutralize sulphuric acid (Weiss et al., 1977). However, sulphate may also be present in other forms, including ammonium disulphate (NH4)3H(SO4)2; the formation of mixed salts with nitrate is unproven, but seems probable under normal atmospheric conditions. The equilibria characterizing mixtures of solids and solutions containing NH4NO3 or mixtures of NH⫹ 4, 2⫺ in equilibrium with NH and HNO at NO⫺ and SO 3 4 3 3 the concentrations typical of the atmosphere have been studied and are reported below: [59] ⫺ NH3(g) ⫹H2SO4(g)⫺ ⫺NH⫹ 4 ⫹HSO4 䉳 䉴 [61] 2⫺ 2NH3(g) ⫹H2SO4(g)⫺ ⫺2 NH⫹ 4 ⫹SO4 ⫺ NH3(g) ⫹HNO3(g)⫺ ⫺NH⫹ 4 ⫹NO3 [62] NH3(g) ⫹H2SO4(g)⫺ ⫺NH4HSO4(s) [63] 2NH3(g) ⫹H2SO4(g)⫺ ⫺(NH4)2SO4(s) [64] NH3(g) ⫹HNO3(g)⫺ ⫺NH4NO3(s) [65] 4NH3(g)⫹2 HNO3(g)⫹ ⫹H2SO4(g)⫺ ⫺(NH4)2SO4⭈2NH4NO3(s) [60] 䉳 䉳 䉴 䉳 䉳 [66] 䉴 䉳 䉳 䉴 䉴 䉴 䉴 5NH3(g)⫹3 HNO3(g)⫹ ⫹H2SO4(g)⫺ ⫺(NH4)2SO⭈3NH 4NO3(s) 4 䉳 䉴 500 dC (equivalent / m3) / dDae (mm) Fig. 4. Distribution of concentration of ions with respect to equivalent diameter. Examining the composition of particulate matter ⫺ indicates the prevalence of NH⫹ 4 and NO3 in the largest size fraction (diameter between 0.1 and 1 mm); this is due to the Kelvin effect which involves the creation of a greater vapour pressure of the volatile species NH3 and HNO3 on strongly curved surfaces. This leads to the volatilization of NH3 and HNO3 from the smallest particles and condensation onto the largest; this phenomenon does not occur for sulphate, which has a low volatility. Fig. 4 shows the distribution of the various species present in particulate matter as a function of the equivalent diameter of the particles themselves. Other components of particulate matter are metals and organic compounds. Most Organic Carbon (OC) is found in the fine fraction of particulate matter. OC derives mainly from the oxidation of combustion products, such as VOCs, and their subsequent condensation, dissolution into the aqueous phase, adsorption (especially onto particles of elementary carbon, EC), or absorption (Seigneur, 2001). The OC found in the particulate matter emitted by motor vehicles contains more than 100 different compounds, including alkanes, benzaldehydes, and Polycyclic Aromatic Hydrocarbons (PAHs), particularly dangerous to human health (Rogge et al., 1993). The organic compounds of greatest medical interest are the polycyclic aromatic hydrocarbons: these form from hydrocarbons with a low molecular mass by pyrosynthesis at temperatures over 500°C. The result is the formation of several aromatic rings condensed into very stable structures which in the atmosphere, as an effect of solar radiation, may be transformed into more dangerous compounds such as nitro-PAHs by reaction with nitric acid and oxidized PAHs by reaction with ozone. The most frequently mentioned PAH is benzo(a)pyrene, which 400 300 200 NH⫹ 4 SO42⫺ NO⫺ 3 Na⫹ Cl⫺ H⫹ 100 0 ⫺2 10 10⫺1 1 10 equivalent diameter Dae (mm) 924 ENCYCLOPAEDIA OF HYDROCARBONS ATMOSPHERIC CHEMISTRY becomes highly carcinogenic through metabolic activation. High concentrations of PAHs are present in the soot generated by the combustion of biomasses and coal, and by the exhausts of diesel and gasoline vehicles. 10.1.3 Atmospheric chemistry in the aqueous phase Basics The total volume of water in the atmosphere is estimated to be about 1.3⭈1013 m3; this water is present in different forms: aerosols, clouds, fog and rain. At the northernmost latitudes, over 30% of the lower troposphere is occupied by cloud bodies. The water content of a cloud is in the order of 0.1-1 g m⫺3 and the size of the droplets forming it depends on the type of cloud; in general, their diameter is greater than 10 mm. Fog has a lower water content, about 0.1 g m⫺3, and smaller droplets. Aqueous aerosols in the atmosphere consist of particles with a broad range of diameters, on the basis of which it is classified as a fine or coarse fraction. A fine fraction contains free water in various forms: dilute aqueous solutions, supersaturated solutions and fine films on insoluble particles. The volume of the drops present in fog and clouds is the liquid medium on which the absorption of reactive trace gases occurs; the majority of these are highly soluble in water. The concentrations of some reactive species may therefore be higher in water than in the surrounding air. This, in conjunction with the high reaction rates of some species in the aqueous phase, leads to the conclusion that the droplets contained in fog and clouds may be extremely efficient reactors for the oxidation of SO2, NO and NO2. An important part of atmospheric chemistry takes place on suspended particles or droplets. The reactions which occur on the surface or inside these particles are described as heterogeneous: they take place on the interface between two phases, i.e. gas-liquid and gas-solid. Those which take place internally rather than on the surface are not heterogeneous in the strict sense of the word, but can be regarded as such considering the volume of air containing the particles. The water content in the atmosphere can be expressed in terms of grams (or of cm3) of water per m3 of air or as the adimensional fraction of volume L (for example, m3 of water per m3 of air). Values of L in different forms of water condensation in the atmosphere are: • Clouds, L⫽10⫺7-10⫺6 • Fog, L⫽5⭈10⫺8-5⭈10⫺7 • Aerosols, L⫽10⫺1-10⫺10 VOLUME III / NEW DEVELOPMENTS: ENERGY, TRANSPORT, SUSTAINABILITY The penetration of atmospheric gases into the suspended droplets involves the following steps: • Transportation of the gas species towards the surface of the drop. • Absorption and transportation across the air-drop interface. • Transportation into the body of the drop. • Reactions inside the drop. The solubility of gases in water is described by Henry’s law, which states that at equilibrium the partial pressure of a gas on a solution containing the same gas is proportional to its concentration in solution. The absorption of a generic species, A, into water can be represented by the following, entirely equivalent, equations: [67] A(g) ⫹H2O⫺ ⫺A⭈H2O 䉳 [68] 䉴 A(g)⫺ ⫺A(aq) 䉳 䉴 The equilibrium between A in the gaseous form and the same species in solution can be expressed by Henry’s constant KH [69] KH ⫽[A(aq)]ⲐpA where pA is the partial pressure of A in the gas phase and [A(aq)] is the concentration of A in the aqueous phase at equilibrium. The measurement units for Henry’s constant are [mol l⫺1 bar⫺1]. Typical values of Henry’s constant for the main atmospheric gases are given in Table 3. The higher the value of this constant, the more soluble the gas in the aqueous phase; however, it is important to remember that some gases subsequently react with the water. Henry’s constant only considers solubility (physical process) and not hydrolysis reactions (chemical process). The effect of equilibria in solution is to increase the quantity of gas passing from the gas phase to the liquid phase in the atmosphere compared to levels predicted based on Henry’s law alone. Given the rapid development of acid-base equilibria, Henry’s law is often extended by defining a Henry’s pseudoconstant KH* which takes into account Table 3. Values of Henry’s constant for the main atmospheric gases Gas KH at 298K (mol l⫺1 bar⫺1) Oxygen 1.3 ⭈10⫺3 Ozone 9.4 ⭈10-3 Nitrogen dioxide 1⭈10⫺2 Carbon dioxide 3.4 ⭈10⫺2 1.24 Sulphur dioxide Ammonia 62 925 ENVIRONMENTAL TECHNOLOGIES all dissolved species. For example, in the case of SO2 we can define: [70] [73] [74] [S(IV)] [SO2(aq)]⫹[HSO⫺ 3] 11144 ⫽ 1211111124 4⫽ KH* S(IV)⫽ p pSO2 SO2 冢 • 冣 K1 K1K2 ⫽KHSO2 1⫹ 11 ⫹ 112 [H ⫹] [H ⫹]2 where K1 and K2 are the first and second dissociation constants of the sulphurous acid. It can be seen from this equation that the value of KH* depends on the pH: the solubility of SO2 decreases as the pH decreases. Despite its high solubility, sulphur dioxide is not found completely dissolved in the tiny water droplets which form clouds. The distribution of a generic species, A, between the gas and aqueous phases in a cloud can be expressed in terms of the ratio of concentrations of A in the two phases per unit volume of air: [71] moles of A in solution111 per litre of air 4 1111121121 11124 ⫽ moles of A in air per litre of air H ⭈p ⭈L K1112 A ⫽1 ⫽KH ⭈R⭈T ⭈L pA ⲐR ⭈T The constant KH is indicated as KH* if the species participates in dissociation equilibria in solution. If KH⭈R⭈T⭈LⰆ1 or KHⰆ1/RTL, species A is present predominantly in the gas phase, whilst the contrary is true if KHⰇ1/RTL. Therefore, if L⫽10⫺6, 1/RTL⯝4⭈10⫺4 mol l⫺1 bar⫺1: if Henry’s constant for a species is less than 4⭈10⫺4 mol l⫺1 bar⫺1 it will be present mainly in the gas phase. If the pH is 4 and L is 10⫺6, the value of H* Ⰶ1/RTL, and SO is therefore KHS(IV)⯝102 gives KS(IV) 2 present mainly as a gas inside clouds. By contrast, for a species such as HNO3, KH*⫽1010 and therefore at equilibrium this species will be present almost entirely in solution. In acidic environments, H2O2 and NH3 are also found mainly in the aqueous phase. The equilibria for the most important species in atmospheric chemistry in the aqueous phase can be described as follows: • carbon dioxide [72] CO2⫹H2O⫺ ⫺H2CO3(aq) 䉳 䉴 ⫺ H2CO3(aq)⫺ ⫺H⫹ (aq) ⫹HCO3 (aq) 2⫺ ⫺ ⫹ HCO⫺ 3 (aq) ⫺H (aq) ⫹CO3(aq) 䉳 䉳 䉴 䉴 sulfur dioxide [76] SO2(g) ⫹H2O⫺ ⫺H2SO3(aq) ⫺ H2SO3(aq)⫺ ⫺H⫹ (aq) ⫹HSO 3(aq) [77] 2⫺ ⫺ ⫹ HSO⫺ 3(aq) ⫺H (aq) ⫹SO3(aq) [75] • 䉳 䉳 䉳 䉴 䉴 䉴 ammonia [78] [79] NH3 ⫹H2O⫺ ⫺NH4OH(aq) ⫺ NH4OH(aq)⫺ ⫺NH⫹ 4(aq) ⫹OH (aq) 䉳 䉳 䉴 䉴 Henry’s constant KH and the equilibrium constants for reactions in the aqueous phase (K⬘ and K⬙) are provided in Table 4. Both in the case of sulphur dioxide and carbon dioxide, the second dissociation constant is much smaller than the first dissociation constant, and can therefore be disregarded at low pH values. The pH of a water droplet in equilibrium with atmospheric CO2 can be calculated by combining the two equilibrium constants referring to the solubility and the first dissociation: [80] H ⫹ [HCO⫺ 3 ] ⫽K ⭈K⬘⭈pCO2 Ⲑ[H ] If the only source of hydrogen ions is the ⫹ dissociation of carbon dioxide, [HCO⫺ 3 ] = [ H ] and therefore: [81] ⫹ H 1/2 [HCO⫺ 3 ] ⫽[H ] ⫽(K ⭈K⬘⭈pCO2) Assuming a partial pressure of CO2 of 340 ppm, this gives a pH of 5.6. This is the pH of rainfall in remote areas; the presence of traces of other compounds may affect acidity: SO2 present at a level of 5⭈10⫺9 bar gives a pH for the solution of 4.6. In fact, for a solution in equilibrium with SO2, it is possible to write an equation similar to that for carbon dioxide: [82] H 1/2 [HSO⫺ 3 ]⫽(K ⭈K⬘⭈pSO2) It therefore seems obvious that even low concentrations of SO2 have a profound effect on pH although the concentration of CO2 in the atmosphere is far higher: this is due to sulphur dioxide’s higher Table 4. Values of Henry’s constant and equilibrium constants in the aqueous phase for NH3, SO2 and CO2 KH at 288K (mol l⫺1 bar⫺1) K⬘(mol l⫺1) Ammonia 90 1.6⭈10⫺5 Sulphur dioxide 5.4 2.7⭈10⫺2 1⭈10⫺7 Carbon dioxide 0.045 3.8⭈10⫺7 3.7⭈10⫺11 Gas 926 K⬙(mol l⫺1) ENCYCLOPAEDIA OF HYDROCARBONS ATMOSPHERIC CHEMISTRY solubility and dissociation constant. These properties give this species a greater acidifying power which may even be intensified by concomitant S(IV)⫺ S(VI) sulphur oxidizing reactions. When SO2 dissolves in an aqueous solution, the ⫺ 2⫺ three resulting species, SO⭈H 2 2O, HSO3 and SO3 , with S(IV), may be oxidized by various species: oxygen (catalyzed by iron and manganese), ozone (the dominant mechanism when pH⬎5) and hydrogen peroxide (the dominant mechanism when pH⬍5). Fig. 5 shows schematically the distribution equilibrium and the subsequent oxidation equilibria of sulphur dioxide. The main oxidation mechanisms are: • ozone 䉴 Sulphur oxidation The oxidation of SO2 in the gas phase is mainly induced by the ⭈OH radical: [83] ⭈OH⫹SO2 (⫹M)⫺ HOSO2 (⫹M) [84] ⫺ ⫺ H2SO4 HOSO⭈⫺ 2 䉴 䉴 䉴 䉴 The ⭈OH radical forms in the atmosphere in the presence of mixtures containing non-methane hydrocarbons and NOx due to the effect of solar radiation and gives rise, through the formation of the aforementioned radical species, to the formation of sulphuric acid. There are various hypotheses regarding the mechanism by which the radical species formed gives rise to H2SO4 (Benson, 1978; Calvert et al., 1978; Davis et al., 1979); this discussion consequently makes use of the simplified equation above. Oxidation in the gas phase induced by the ⭈OH radical is characterized by formation rates greater than 1% an hour; studies conducted both in the field and in the laboratory have recorded values for the conversion rate higher than those predictable on the basis of gas phase chemistry alone. This leads to the conclusion that reactions in the aqueous phase may be important sources of sulphate and in some cases may even be more important than those in the gas phase. [85] • ⫺ 2⫺ HSO⫺ 3(aq) ⫹OH ⫹O3⫺ SO4 ⫹H2O ⫹O2 䉴 hydrogen peroxide [86] ⫺ ⫺ HSO⫺ 3(aq) ⫹H2O2(aq) ⫺SO2OOH ⫹H2O [87] SO2OOH⫺ ⫹H⫹⫺ H2SO4 • 䉳 䉴 䉴 oxygen [88] Fe, Mn 2⫺ ⫹H⫹ HSO⫺ ⫺ SO4(aq) 3(aq) ⫹1Ⲑ2O2(aq)⫺ (aq) 䉴 Ozone reacts very slowly with SO2 in the gas phase, whereas the reaction is fast in the aqueous phase. The most plausible mechanism is the ionic mechanism shown above and proposed by Maahs (1983). Radical mechanisms have also been proposed whose contribution to the oxidation of S(IV) is not quantifiable with any degree of certainty. These may take place via a radical intermediate such as ⭈OH (Hoignè and Bader, 1975; Penkett et al., 1979): SO2(g) Fig. 5. Sulphur dioxide distribution and oxidation equilibria. (1) transport to droplet surface (2) transport across air-water interface SO2(interface) SO2⭈H2O HSO⫺ 3 (3) establishment of S(IV) equilibria SO2⫺ 3 (4) S(VI) transport into bulk phase S(IV) (5) VOLUME III / NEW DEVELOPMENTS: ENERGY, TRANSPORT, SUSTAINABILITY oxidation 927 ENVIRONMENTAL TECHNOLOGIES [89] [90] [91] ⫺ HSO⫺ 3 ⫹⭈OH⫺ H2O ⫹SO3 ⫺ SO⫺ 3 ⫹O2⫺ SO5 ⫺ ⫺ 2SO5 ⫺ SO4 ⫹SO2⫺ 4 ⫹O2 䉴 䉴 䉴 Oxidation induced by H2O2 is relatively independent of pH and this is due to the fact that the rate constant of the reaction and the solubility of S(IV) show opposing trends with respect to pH. Other species are affected in different ways by variations in pH: in the case of ozone the reaction rate increases by one order of magnitude when passing from pH 1 to pH 3, whereas the reaction rate of oxygen catalyzed by iron increases by two orders of magnitude. The dependence of rate and the solubility of S(IV) on pH means that the reaction with ozone is only important when pH⬎5. Research has concentrated particularly on reactions in the aqueous phase catalyzed by transition metals: the most important catalysts are iron (in the form Fe3⫹) and manganese (in the form Mn2⫹). Some studies have been carried out on the possible catalytic action of other metals such as Cu2⫹ and Co2⫹; however, the reactions catalyzed by the latter are characterized by very low reaction rates under typical ambient conditions. The rate of the reaction catalyzed by Fe(III) depends on various parameters, such as pH, ionic force and temperature, and is affected by the presence of some anions (such as SO42⫺) and cations (Mn2⫹) in solution. The mechanisms of these reactions have long been debated. A radical mechanism has been suggested (Hoffmann and Boyce, 1983; Hoffmann and Jacob, 1984): • initiation (n⫺1)⫹⫹SO⭈⫺ [92] Mn⫹⫹SO2⫺ 3 ⫺M 3 䉴 • propagation [93] ⫺ SO⭈⫺ 3 ⫹O2⫺ SO⭈5 [94] 2⫺ 2⫺ ⫺ SO⭈⫺ 5 ⫹SO3 ⫺ SO5 ⫹SO⭈3 • 䉴 䉴 metal-sulphite complex followed by bonding with oxygen: 2⫺ ⫺ Mn2⫹⫹2SO2⫺ 3 ⫺Mn(SO3)2 2⫺ ⫺ [100] Mn(SO3)2⫺ 2 ⫹O2 ⫺Mn(SO3)2O2 2⫹ 2⫺ ⫺ [101] Mn(SO3)2O2⫺ 2 ⫺Mn ⫹2SO4 [99] 䉳 䉴 䉳 䉳 䉴 䉴 Mechanisms that are even partially photochemical have been suggested, based on the observation (Lunak and Veprek-Siska, 1976) that the oxidation of S(IV) in homogeneous aqueous systems for wavelengths above 300 nm does not occur unless Fe3⫹ is present. Photooxidation has been attributed to the absorption of light by a Fe3⫹- S(IV) complex. In the case of iron, this is present in solution in the soluble and solid form: [102] [Fe]tot ⫽[Fe]sol ⫹[Fe(OH)3] Soluble iron is found in aqueous solution in various ionic forms: [103] [Fe]sol ⫽[Fe3⫹] ⫹[FeOH2⫹] ⫹[Fe(OH)⫹2 ] ⫹ ⫹[Fe2(OH)24⫹] The equilibrium between iron in the soluble form and solid iron is as follows: [104] Fe(OH)3 ⫹3H⫹⫺ ⫺Fe3⫹ ⫹3H2O 䉳 䉴 As a result, the relative quantities of the different forms of iron in aqueous solution are strongly influenced by pH. With pH values of 4.5, [Fe3⫹]⯝3⭈10⫺11; above this pH value the soluble form of iron decreases significantly. Another catalyst for these oxidation reactions is manganese: attempts have been made to determine which of the two metals is the main cause of SO2 oxidation, but to date their relative importance in the reaction has not been clarified. It is likely that the two catalysts act in synergy: an increase in the reaction rate has been observed in the presence of both ions, higher than would be expected on the basis of the sum of their respective catalysis oxidation [95] 2⫺ 2⫺ SO2⫺ 5 ⫹SO3 ⫺ 2SO4 䉴 • termination 2⫺ [96] 2SO⭈⫺ 3 ⫺ S2O6 ⫺ 2⫺ [97] SO⭈⫺ 3 ⫹SO⭈5 ⫺ S2O6 ⫹O2 ⫺ ⫺ [98] SO5 ⫹SO5 ⫺ S2O82⫺ ⫹O2 䉴 Table 5. Concentrations (expressed in molarity, M) of iron and manganese in various aqueous matrices present in the atmosphere 䉴 䉴 This hypothesis was later abandoned since it clashed with the kinetic order of reaction of the reagents and was replaced by two alternative mechanisms: ionic and photochemical. The ionic mechanism, initially proposed by Bassett and Parker (1951), involves first the formation of a 928 AQUEOUS MATRIX Mn Fe Mist 10⫺7-10⫺4 10⫺4-10⫺3 Clouds 10⫺8-10⫺5 10⫺7-10⫺4 Rain 10⫺8-10⫺6 10⫺8-10⫺5 Fog 10⫺7-10⫺5 10⫺6-10⫺4 ENCYCLOPAEDIA OF HYDROCARBONS ATMOSPHERIC CHEMISTRY rates: the removal of S(IV) is about 3–10 times faster if there is synergy between the two species (Martin, 1984). Table 5 quantifies the presence of these two species in the various forms of water condensation in the atmosphere. Temperature has the opposite effect on these catalyzed reactions compared to other oxidation reactions: the formation of sulphate via these mechanisms decreases as temperature decreases. In fact, the presence of the catalysts does not vary with temperature, as is the case for the other oxidizing species found in the gaseous form, whilst the high activation energies are affected by decreases in temperature. Oxidation by Mn2⫹ is inversely influenced by pH with respect to Fe3⫹: whereas in the case of iron the reaction rate decreases as pH decreases in the range 0-4, the opposite occurs for manganese in the range 0-3. Fig. 6 shows the effect of pH on the various types of oxidation. It can be observed that the dominant mechanism for the formation of sulphate at pH lower than 4-5 is that induced by hydrogen peroxide. In contrast, when pH⭓5 oxidation by ozone is 10 times faster than that by H2O2. Oxidation catalyzed by metals is important when the pH is high. Assuming a water content of 1 g m⫺3 within a cloud, the rate of oxidation induced by H2O2 exceeds 100% an hour, whereas the rates of oxidation reactions catalyzed by iron and manganese are lower than 1% an hour when pH ⬍4.5. 104 O3, 50 ppb 103 H2O2, 1 ppb rate of S(IV) oxidation (%/h) 102 Fe, 3ⴢ10⫺7mol/l 101 Mn, 3ⴢ10⫺8mol/l The oxidizing power of nitrogen oxides with respect to S(IV) differs depending on the species under consideration. NO and HNO3 induce reactions which are too slow to contribute effectively to oxidation; HONO, on the other hand, has extremely low atmospheric concentrations (1-8 ppb) and, despite its high Henry’s constant (KH⫽49 mol l⫺1 bar⫺1), does not reach sufficient concentrations in solution to represent an important oxidizing agent. NO2, whose Henry’s coefficient is KH⫽1⭈10⫺2 mol l⫺1 bar⫺1, by contrast, is a relatively insoluble gas; however, it has been shown (Schwartz, 1984) that the 2⫺ reaction rates of NO2 with HSO⫺ 3 and SO3 are sufficiently high to make its oxidizing action important. Additionally, it has been shown that the reaction: ⫹ ⫺ 2⫺ [105] 2NO2⫹HSO⫺ 3 ⫺ 3H3O ⫹2NO2 ⫹SO4 䉴 may be sufficiently fast at high pH levels (Lee and Schwartz, 1983; Lee, 1984). Oxidation of nitrogen In addition to oxidation from S(IV) to S(VI), another possible oxidation process leading to the acidification of the water present in clouds or rain is that induced by NO2 and NO and giving rise to nitric and nitrous acid: ⫺ ⫺ [106] 2NO2(g) ⫹H2O(l)⫺ 2H⫹ (aq) ⫹NO 3(aq) ⫹NO 2(aq) 䉴 ⫺ [107] NO(g) ⫹NO2(g) ⫹H2O(l)⫺ 2H⫹ (aq) ⫹2NO 2(aq) 䉴 However, these reactions do not contribute significantly to acidification at the concentrations of nitrogen oxides normally present in the atmosphere: the first of the two reactions (Schwartz, 1984) is very slow, both due to the low solubility of NO2 and to the second order dependence of the reaction rate on the concentration of NO2. The formation mechanism for nitric acid in air remains primarily the same as the gas phase induced by the free radical ⭈OH: [108] ⭈OH ⫹NO2 (⫹M)⫺ HONO2 (⫹M) 䉴 1 C, 10⫺2g/l 10⫺1 HNO2, 1 ppb NO2, 2 ppb 10⫺2 10⫺3 10⫺4 10⫺5 Analysing the composition of precipitation and the surrounding air masses nevertheless shows that oxidation in the aqueous phase also produces significant quantities of nitric acid in the atmosphere (Lazrus et al., 1983; Misra et al., 1985). A mixed gas-liquid mechanism for the production of nitric acid in droplets has been proposed by Heikes and Thompson (1983): [109] O3 ⫹NO2⫺ O2 ⫹NO3 䉴 0 1 2 3 4 5 6 pH Fig. 6. Effect of pH on various sulphur(IV) oxidation reactions. VOLUME III / NEW DEVELOPMENTS: ENERGY, TRANSPORT, SUSTAINABILITY [110] NO3 ⫹NO2 (⫹M)⫺ N2O5 (⫹M) 䉴 N2O5 generated in this way in the gas phase may be incorporated into droplets or onto the surface of aqueous aerosol, and hydrolyze to HNO3: 929 ENVIRONMENTAL TECHNOLOGIES ⫺ [111] N2O5 ⫹H2O⫺ 2H⫹ (aq) ⫹2NO 3(aq) 䉴 However, a definitive evaluation of the contribution of this mechanism to the concentration of HNO3 in the liquid phase of the atmosphere is not yet possible. Special attention has recently been devoted to reactions in the aqueous phase involving species generated by photochemical mechanisms: inside cloud bodies there may be sufficient light for the production of hydroxyl and hydroperoxide radicals. Hydroxyl radicals react with nitrogen oxides to form nitrous and nitric acid: ⫺ [112] NO⫺ 2(aq) ⫹⭈OH(aq)⫺ NO2(aq) ⫹OH (aq) 䉴 [113] NO(aq) ⫹⭈OH(aq)⫺ HNO2(aq) 䉴 [114] NO2(aq) ⫹⭈OH(aq)⫺ HNO3(aq) 䉴 Other potential reactions leading to the formation of nitric acid are those suggested by Heikes and Thompson (1983) and Platt et al. (1984): the nitrate radical absorbed in solution is hydrolyzed to form nitric acid. This may occur starting from the radical itself with the extraction of a hydrogen atom from an organic compound: [115] NO⭈⫹RH⫺ R⭈⫹HNO3 3 䉴 The reaction with H2O2, however, does not appear to be important: ⫹ [116] HONO ⫹H2O2⫺ NO⫺ 3 ⫹H3O 䉴 since it has been demonstrated (Lee, 1984) that this reaction is too slow. In the absence of clouds and in the presence of solar radiation, HNO3 is generated in the gas phase by the reaction of NO2 with ⭈OH radicals at a rate of about 20-30% per hour, whilst H2SO4 is generated by this pathway far less effectively. By contrast, in the aqueous phase oxidation reactions involving H2O2, O3 and metal catalysts are able to produce H2SO4 in solution at rates of up to 100% an hour. The chemico-physical properties of these two acid species are fundamentally different: nitric acid is far more volatile and tends to remain in the atmosphere in the gaseous form, whilst sulphuric acid has a relatively low vapour pressure (⬍10⫺7 bar) and therefore tends to be present inside particles. Both may react with basic species. These reactions essentially occur with the ammonia present to form ammonium nitrate and ammonium sulphate: [117] HNO3 ⫹NH3⫺ ⫺NH4NO3 䉳 䉴 [118] H2SO4 ⫹2NH3⫺ ⫺(NH4)2SO4 䉳 䉴 These two species are the main cause of diminishing visibility in the case of photochemical 930 smog. Ammonium nitrate is found in the solid form if the temperature is below that of deliquescence; if relative humidity is high, it is present in the aqueous phase. However, nitric acid may return to the gas phase even after forming ammonium salts. Importance of aqueous phase equilibria in the formation of acid rain Formed in the atmosphere by the mechanisms described above in the aqueous and gas phases, the acids under examination may be deposited on the Earth’s surface by two main mechanisms: dry deposition and wet deposition. The distinction is made based on the phase in which the pollutant is found when it hits the surface: dry deposition involves gaseous pollutants or small particles, whilst wet deposition involves pollutants present in the droplets inside fog, clouds and rain. It should be specified that this classification only considers the transportation mechanism, and not the nature of the surface (whether or not it is wet or if it presents a liquid film). Given the variable nature of precipitation it is difficult to make quantitative calculations of the extent of wet deposition of a pollutant species. The deposition rate of a pollutant is given approximately by the product l⭈C, where C is the concentration of the pollutant and l is the scavenging coefficient proportional to the intensity of the precipitation itself. Dry deposition, in contrast, is characterized by the deposition rate Vg, defined as: [119] Vg ⫽FⲐ[S] where F is the flux of species S towards the surface and [S] is the concentration of the same species at a reference altitude h. As far as wet deposition by precipitation is concerned, the sulphate and nitrate deposited on exposed surfaces may be the result not only of oxidation reactions in the aqueous phase, but also of the inclusion of aerosol particles containing these species inside the cloud or of the scavenging of aerosol particles beneath the cloud itself. If these inclusion and scavenging mechanisms predominate, acidification will be determined by the parameters governing the formation of nitrates and sulphates in the gas phase: solar radiation, the concentration of NOx and hydrocarbons. If formation inside the droplets by oxidation of SO2 and NO2 prevails, however, the parameters determining acidity will be the concentrations of oxidants such as H2O2 and O3. Comparing the composition of interstitial aerosols (a fraction of the atmospheric aerosol which, in the ENCYCLOPAEDIA OF HYDROCARBONS ATMOSPHERIC CHEMISTRY presence of a cloud, remains the same and is not removed by the water droplets present inside the cloud) and that of clouds provides information on the source of acidity inside the clouds themselves. Daum and colleagues (1983) have shown that for interstitial aerosols the ratio [H⫹]/[NH⫹ 4 ] is less than 1, whereas the aerosol contained in clouds has a ratio greater than 1. Similar conclusions have been reached by other research groups (Lazrus et al., 1983; Harrison and Pio, 1983): the higher acidity of the aerosols in cloud bodies with respect to that of the surrounding atmosphere agrees well with the formation mechanisms of acidic species in the aqueous phase described above. It can be concluded that the formation of sulphuric acid inside cloud aerosols provides the largest contribution to the formation of sulphate in acid rain. 10.1.4 Depletion of stratospheric ozone The stratosphere is that region of the Earth’s atmosphere which stretches from about 15 to 50 km above the troposphere. The temperature is almost constant in the lowest layer, whilst it tends to increase gradually in the upper half. The temperature pattern in the stratosphere is regulated by variations in the concentration of ozone inside this layer: by absorbing solar radiation in the ultraviolet band ozone molecules convert solar energy into kinetic energy, helping to heat the stratosphere. The average global concentration of stratospheric ozone varies with altitude; at an altitude of 15 km it is in the order of 0.5 parts per million (ppm) by volume, at around 35 km it increases up to 8 ppm and then decreases to 3 ppm in the high stratosphere (45 km). The thickness of the ozone layer above a certain geographic area or its average global value can be conventionally expressed in Dobson Units (DU), in other words with a height of 0.01 mm; the ozone would be at this thickness if it were the only component of the atmosphere and if it were at a pressure of 1 bar and at a temperature of 0°C. On average the height of the ozone layer may vary with latitude between 250 and 400 DU; its average global value is about 300 DU. The concentration of ozone in the various regions of the stratosphere is the result of a dynamic formation and destruction process. Ozone forms at an altitude of about 30 km, where ultraviolet solar radiation with a wavelength shorter than 242 nm slowly dissociates the oxygen molecules into atomic oxygen: [121] O ⫹O2 ⫹M⫺ O3 ⫹M 䉴 In turn, the ozone molecules absorb the high energy photons present in solar radiation, with wavelengths ranging from 240 to 320 nm. A direct consequence of this absorption is the dissociation of ozone into an oxygen molecule and an excited oxygen atom, O(1D), according to the reaction: [122] O3 ⫹hv⫺ O2 ⫹O 䉴 This absorption process means that a significant portion of ultraviolet solar radiation does not reach the Earth’s surface: this has made the development of life possible on our planet. The photolysis of ozone is not a genuine destruction mechanism because virtually all the oxygen atoms produced by this reaction combine rapidly with oxygen molecules to form ozone again. This mechanism entails the conversion of solar energy into thermal energy, especially in the upper part of the stratosphere. The presence of ozone is thus the cause of the temperature inversion characterizing the upper belt of the stratosphere. The current distribution of stratospheric ozone is mostly determined by transportation processes. Ozone is produced mainly at the tropics at an altitude of between 25 and 35 km but, as a result of the movements of air masses, its highest concentrations are found near the poles at an altitude of about 15 km. Various destruction mechanisms contribute to balancing out ozone formation processes (reactions [120] and [121]). An example is the reaction of ozone with oxygen atoms to form molecular oxygen: [123] O3 ⫹O⫺ O2 ⫹O2 䉴 The above reactions are known as Chapman reactions and have formed the basis for the study of stratospheric ozone. Chapman’s scheme only estimates the loss of ozone through natural destruction and does not consider the transportation of ozone onto the Earth’s surface which contributes a further 0.5%. About 10% of destruction processes can be attributed to catalytic cycles involving species which contain hydrogen: free hydrogen atoms (H), hydroxyl (⭈OH) and hydroperoxide (HO⭈) 2 radicals give the same results as reaction [123]. For example: ⭈OH ⫹O2 O ⫹HO⭈⫺ 2 HO⭈⫹O3⫺ HO⭈⫹O 2 2 11111111122 O3 ⫹O⫺ O2 ⫹O2 These species containing hydrogen are generated by the reaction which normally occurs between water vapour and methane and the excited oxygen atoms O (1D) from the photolysis of ozone. 䉴 䉴 䉴 [120] O2 ⫹hv⫺ O ⫹O 䉴 The oxygen atoms rapidly combine with molecular oxygen to form ozone; in the presence of a third inert molecule, M, the following reaction may take place: VOLUME III / NEW DEVELOPMENTS: ENERGY, TRANSPORT, SUSTAINABILITY 931 ENVIRONMENTAL TECHNOLOGIES A significant contribution, about 70%, to the process of ozone destruction is supplied by a catalytic cycle involving NO and NO2, the main reactions of which are: O⫹NO2⫺ NO⫹O2 NO⫹O3⫺ NO2⫹O2 11111111122 O3⫹O⫺ O2⫹O2 The main source of NOx in the stratosphere is the oxidation of the nitrous oxide (N2O) produced by bacteria in the land and water. Although most nitrous oxide is converted into N2 and O by ultraviolet light, about 1% reacts with the excited oxygen atoms O (1D) generated by the action of ultraviolet radiation on ozone to form nitrogen oxide and begin the cycle: 䉴 Table 6. Halogenated compounds present in the troposphere Name Chemical formula CFC-11 Trichlorofluoromethane CFCl3 CFC-12 Dichlorofluoromethane CF2Cl2 CFC-113 Trichlorotrifluoroethane C2F3Cl3 CFC-114 Dichlorotetrafluoroethane C2Cl2F4 CFC-115 Chloropentafluoroethane C2ClF5 Halon-1211 Bromochlorofluoromethane CF2ClBr Halon-1301 Bromotrifluoromethane CF3Br Compound 䉴 䉴 [124] O (1D) ⫹N2O⫺ NO ⫹NO 䉴 The NOx molecules, in turn, are removed by the formation of nitric acid (HNO3) in the reaction ⭈OH-NO2 which occurs in the lowest layer of the stratosphere. Atmospheric currents transport the nitric acid towards the troposphere, where it is removed by precipitation; in the same way, the nitrous oxide from the troposphere is transported upwards and destroyed. The destruction of ozone may also be catalyzed by substances other than HOx e NOx, in particular by chlorine (Cl) and bromine (Br) atoms and their respective oxides (ClO and BrO). The year 1930 saw the beginnings of the industrial production of chlorofluoromethanes and chlorofluoroethanes, known by the name freon, which became widely used as liquid refrigerants, solvents and propellants for aerosol cans. In 1975, two researchers, F.S. Rowland and M.J. Molina, published an article stating that in the stratosphere freon could induce radical chain reactions, with negative effects the on the natural equilibrium of ozone. An example of the reactions taking place is as follows: • initial stage [125] CF2Cl2 ⫹hv⫺ CF2Cl⭈⫹Cl 䉴 • propagation stage [126] O ⫹ClO⭈⫺ Cl⭈⫹O2 䉴 [127] Cl⭈⫹O3⫺ ClO⭈⫹O2 䉴 In the initial stage, ultraviolet light causes the homolytic cleavage of a C⫺Cl bond of the freon. The chlorine radical may induce the propagation of a chain reaction leading to the destruction of ozone molecules. Again in 1976, a study by the National Academy of Sciences (NRC, 1976) confirmed Rowland and Molina’s predictions and in January 1978 the use of freon in aerosol cans was banned in the United States. The main chlorine compounds present in the troposphere (Table 6) are methyl chloride (CH3Cl), a 932 tiny proportion of which is of industrial origin, the man-made chlorofluoromethanes CFCl3 (CFC-11) and CF2Cl2 (CFC-12), and carbon tetrachloride (CCl4) generated by both natural and man-made sources. Less important man-made sources include trichloroethylene (CCl2⫽CHCl) and the substances which have replaced it, methyl chloroform (CH3CCl3) and trichlorotrifluoroethane (C2F3Cl3), CFC-113. From the point of view of their effects on the stratosphere, the key species are the chlorofluorocarbons CFCl3 (CFC-11) and CF2Cl2 (CFC-12) whose concentrations in the atmosphere increased by 37% and 31% respectively between 1976 and 1981. It is thought that the residence times of CFC-11 and CFC-12 are about 60 and 110 years respectively; measurements indicate a minimum life-time for CFC-11 of 40 years. A few years after their commercialization, these chlorine compounds spread through the troposphere, and their concentration subsequently began to increase slowly in the stratosphere as well. Chlorofluorocarbons (CFC-11, CFC-12, CFC-113) are highly inert compounds in the troposphere and in the lower layers of the stratosphere, but when transported to altitudes of 25-50 km they are decomposed by ultraviolet radiation with wavelengths shorter than about 200 nm, with the resulting formation of chlorine atoms. The chlorine atoms subsequently participate in the catalytic cycle of ClOx. This cycle may be interrupted by the conversion of the highly reactive forms Cl and ClO into less reactive forms which do not destroy ozone. The chlorine atoms are deactivated by reaction with methane to form HCl: [128] Cl⫹CH4⫺ HCl⫹CH3 䉴 ENCYCLOPAEDIA OF HYDROCARBONS ATMOSPHERIC CHEMISTRY which acts as a temporary reservoir for the active chlorine species in the stratosphere. The chlorine atoms are regenerated by the reaction between HCl and ⭈OH radicals: [129] ⭈OH ⫹HCl⫺ H2O ⫹Cl 䉴 The destruction and regeneration processes of the active chlorine species, ClOx , may occur several times before the chlorine is completely removed from the stratosphere. The most important removal mechanism is the transportation of HCl from the stratosphere to the upper troposphere, from which it is removed by the action of rain, as occurs for the removal of nitric acid from the NOx cycle. The time scale from the point at which the chlorofluorocarbons are emitted at ground level to the point at which their chlorine atoms, in the form of HCl, are removed from the atmosphere by rain is in the order of several decades; variations in the emission fluxes of chlorofluorocarbons thus manifest themselves in the stratosphere after many years. Ozone destruction reactions may also be catalyzed by bromine atoms. It is thought that stratospheric bromine is 50 times more active than Cl in the processes leading to the destruction of ozone, and that it is responsible for 20% of the ozone hole over the Antarctic and for a greater proportion of that over the Arctic. The most important transportation vehicle for bromine is methyl bromide (CH3Br); the photolysis of this compound leads to the formation of the Br radical, responsible for the demolition of O3: [130] CH3Br ⫹hv⫺ ⭈CH3 ⫹⭈Br In order to replace chlorofluorocarbons, other substances have been synthesized for use as refrigerants and aerosol propellants: hydrochlorofluorocarbons (HCFCs) and hydrofluorocarbons (HFCs). The presence of at least one hydrogen atom in each molecule means that these are oxidized in the troposphere by reactions with ⭈OH radicals: [134] ⭈OH ⫹CHClxFy⫺ H2O ⫹CClxFy⫺ products 䉴 䉴 The presence of hydrogen thus allows for a faster degradation of the substance in the atmosphere, and therefore a lower environmental impact (HCFCs and HFCs do not reach the stratosphere). The Ozone Depleting Potential (ODP) has been introduced to combat the harmful effects of the chlorofluorocarbons and the substances which have replaced them. For any given halocarbon, the ODP is defined as the ratio of stratospheric ozone destroyed by the emission of 1 kg of that compound to the ozone destroyed by the emission of 1 kg of CFC-11. A substance’s ozone depleting potential thus provides a measure of the impact (in comparison to that of CFC-11, considered the standard reference compound with an ODP of 1.00) of the emission of 1 kg of that substance in terms of the destruction of stratospheric ozone. As such, the impact of a substance on the ozone layer is given by its capacity to destroy ozone and the quantity of total emissions. Table 7 shows the ODP values and life-times in the atmosphere of the main compounds. 䉴 [131] Br ⫹O3⫺ BrO⭈⫹O2 䉴 [132] BrO⭈⫹O⫺ ⭈Br ⫹O2 䉴 Little is known about methyl bromide’s sources and cycle, unlike chlorofluorocarbons. This gas is used as a fumigant for the antiparassitic treatment of land and agricultural products and as a raw material for products of chemical synthesis. Together, natural sources (mainly marine biological activity; Lovelock, 1975) and man-made sources emit about 100 kt a year into the environment. Considering the emission fluxes and a life-time of about 2 years, the average global concentration of methyl bromide is on the order of 10 ppt. A particularly interesting aspect of the chemistry of stratospheric bromine is the possible synergic interaction between its cycle and that of chlorine, through the reaction: [133] BrO ⫹ClO⫺ Br ⫹Cl ⫹O2 Table 7. ODP values and life-times of the main halogenated compounds present in the atmosphere (WMO/United Nations Environment Programme, 1994) ODP Life-time in the atmosphere (years) CFC-11 1.00 60 CFC-12 0.82 110 CFC-113 0.90 90 CFC-114 0.85 200 CFC-115 0.4 400 HCFC-22 0.04 13.3 HCFC-123 0.014 1.4 Halon-1211 5.1 20 Halon-1301 12 85 Compound 䉴 Yung and colleagues (1980) suggest that the interaction between ClOx and BrOx may lead to an increase in the ozone destruction process in the lower layers of the stratosphere. VOLUME III / NEW DEVELOPMENTS: ENERGY, TRANSPORT, SUSTAINABILITY 933 ENVIRONMENTAL TECHNOLOGIES Other reactions may occur in the heterogeneous phase on the surface of the particles present in Polar Stratospheric Clouds (PSCs), consisting of an icy mixture of nitric acid and water, generated at temperatures below 195K (⫺78°C); these clouds, pearly white in colour, can frequently be seen over the Antarctic. HCl molecules aggregate onto the ice crystals, becoming part of the crystalline structure and leading to the formation of a solid phase catalyst. The catalyst reacts with chlorine nitrate to release chlorine in the gaseous state; this accumulates in the stratosphere and participates in the ozone destruction process: [135] ClONO2 ⫹HCl⫺ Cl2 ⫹HNO3 䉴 The nitric acid remains bonded to the surface of the particles and therefore reacts with NO2, which is no longer able to neutralize ClO. In this way, the level of active chlorine atoms is kept constant. The low temperatures reached in polar Antarctic regions are associated with low pressure conditions leading to the formation of a polar vortex which mixes troposphere and stratosphere whilst simultaneously preventing the entry of external air masses. The same low temperatures encourage the formation of PSCs in the Antarctic region, a phenomenon of minor importance in the Arctic, where temperatures are about 10-15°C higher. It appears that aerosol particles containing sulphuric acid may present surface conditions that accelerate the ozone destruction process. This was shown clearly following the high emissions into the stratosphere of sulphur compounds in particulate and gaseous form by explosive volcanic eruptions (an example is the eruption in 1991 of Mount Pinatubo in the Philippines). The release of large amounts of SO2 leads to the formation of aerosol through the reaction: the water vapour present in small quantities in the stratosphere to form aerosol droplets. Volcanoes are only one source of the gaseous sulphur present in the stratosphere; another, of biogenic origin, releases carbon disulphide. The levels of carbon disulfide present in the stratosphere are extremely low, but it may be transferred there across the tropopause, unlike other sulphur compounds such as hydrogen sulphide and sulphur dioxide, apparently too reactive to leave the troposphere. The high concentrations of particulate matter containing sulphate (in the form of both SO42⫺ and H2SO4) are shown in Fig. 7. The diagram shows that carbonyl sulphide (OCS) plays an important role in the chemistry of the sulphur compounds present in the atmosphere. It can be oxidized to sulphuric acid: [140] OCS ⫹⭈OH⫺ CO2 ⫹HS 䉴 [141] HS ⫹⭈OH⫺ SO ⫹H2 䉴 [142] SO ⫹⭈OH⫺ SO2 ⫹H 䉴 It has been estimated that over 50% of the carbonyl sulphide present in the atmosphere is man made and is emitted by combustion processes and the treatment of fossil fuels. Some models suggest that one of the most significant consequences of ozone depletion will be the cooling of the lower part of the stratosphere. It is thought that there is a positive feedback mechanism by which the loss of ozone cools the air, encouraging the formation of stratospheric polar clouds which in turn 40 H2SO4 30 [136] ⭈OH ⫹SO2⫺ HSO3 which may considerably decrease the concentrations of ⭈OH radicals in the upper part of the atmosphere, with notable consequences. The subsequent reactions involving HSO3 are not fully understood, but a potential further step might be: [137] HSO3 ⫹⭈OH⫺ H2SO4 䉴 altitude (km) 䉴 SO42⫺ SO2 20 tropopause OCS 10 CS2 similarly H2S and (CH3)2S Alternatively, two other reactions could be suggested: [138] HSO3 ⫹O2⫺ HSO5 䉴 [139] HSO3 ⫹O2⫺ HO⭈⫹SO 3 2 䉴 The oxidation of SO2 of volcanic origin in the atmosphere takes place very slowly. The sulphuric acid generated in this way may subsequently condense on 934 0 10⫺12 10⫺11 10⫺10 10⫺9 concentration (molar fraction) Fig. 7. Concentration of the main sulphur compounds as a function of altitude. ENCYCLOPAEDIA OF HYDROCARBONS ATMOSPHERIC CHEMISTRY contribute to the lowering of ozone levels. It is obvious that the formation of PSCs is not limited to polar vortices, but may occur inside stratospheric jet streams at temperate latitudes. All this suggests that heterogeneous processes involving aerosols containing ice particles, sulphuric acid and sulphur compounds are important components of the chemistry of the stratosphere. References Bassett H., Parker W.G. (1951) Oxidation of solfurous acid, «Journal of the Chemical Society», 1540-1560. Benson S.W. (1978) Thermochemistry and kinetics of sulfur containing molecules and radicals, «Chemical Reviews», 78, 23. Calvert J.G., Stockwell W.R. (1984) Mechanism and rates of the gas-phase oxidations of sulphur dioxide and nitrogen oxides in the atmosphere, in: Calvert J.G. (editor) SO2, NO and NO2 oxidation mechanisms. Atmospheric conditions, Boston (MA), Butterworth. Calvert J.G. et al. (1978) Mechanism of the homogeneous oxidation of sulfur dioxide in the troposphere, «Atmospheric Environment», 12, 197-226. Clement C.F., Ford I.J. (1996) Seasonal variation of aerosol particles, «Journal of Aerosol Science», 27, 39-40. Crutzen P.J. (1988) Tropospheric ozone: an overview, in: Isaksen I.S.A. (edited by) Tropospheric ozone, Dordrecht, Reidel, 3-32. Daum P.H. et al. (1983) Studies of the gas- and aqueous-phase composition of stratiform clouds, in: Pruppacher H.R. et. al. (coordinators) Precipitation scavenging, dry deposition and resuspension. Proceedings of the 4th International conference, Santa Monica (CA), 29 November-3 December 1982, New York, Elsevier, 2v.; v.II, 795-804. Davis D.D. et al. (1979) Sulfur dioxide oxidation via the hydroxil radical: atmospheric fate of HSOx radicals, «Geophysical Research Letters», 6, 113-116. Harrison R.M., Pio C.A. (1983) An investigation of the atmospheric nitric acid-ammonia-ammonium nitrate equilibrium relationship in a cool, humid climate, «Tellus. Series B: Chemical and Physical Meteorology», 35B, 155-159. Heikes B.G., Thompson A.M. (1983) Effects of heterogeneous processes on nitrogen trioxide, nitrous acid, and nitric acid chemistry in the troposphere, «Journal of Geophysical Research. C: Oceans and Atmosphere», 88, 10883-10895. Hoffmann M.R., Boyce S.D. (1983) Atalytic autoxidation of aqueous sulfur dioxide in relationship to atmospheric system, «Advances in Environmental Science and Technology», 12, 147-189. Hoffmann M.R., Jacob D.J. (1984) Kinetics and mechanisms of catalytic oxidation of dissolved sulfur dioxide in aqueous solution: an application to nighttime fog chemistry, in: Calvert J.G. (editor) SO2, NO and NO2 oxidation mechanisms. Atmospheric considerations, Boston (MA), Butterworth, 101-172. Hoignè J., Bader H. (1975) Ozonation of water. Role of hydroxyl radicals as oxidizing intermediates, «Science», 190, 782-784. VOLUME III / NEW DEVELOPMENTS: ENERGY, TRANSPORT, SUSTAINABILITY Jacobson M.Z. (1998) Studying the effects of aerosols on vertical photolysis rate coefficient and temperature, «Journal of Geophysical Research. Atmospheres», 103, 10593-10604. Jacobson M.Z. (1999) Isolating nitrated and aromatic aerosols and nitrated aromatic gases as sources of ultraviolet light absorption, «Journal of Geophysical Research. Atmospheres», 104, 3527-3542. Kelly N.A., Gunst R.F. (1990) Response of ozone to changes in hydrocarbon and nitrogen oxide concentrations in outdoor smog chambers filled with Los Angeles air, «Atmospheric Environment. Part A: General Topics», 24A, 2991-3005. Kikas U. et al. (2001) A case study of the impact of boundary layer aerosol size distribution on the UV irradiance, «Atmospheric Environment», 35, 5041-5051. Lazrus A.L. et al. (1983) Acidity in air and water in a case of warm frontal precipitation, «Atmospheric Environment», 17, 581-591. Lee Y.-N. (1984) Atmospheric aqueous-phase reactions of nitrogen species, in: Gas-liquid chemistry of natural waters, Brookhaven (NY), Brookhaven National Laboratory, 2v.; v.I. Lee Y.-N., Schwartz S.E. (1983) Kinetics of oxidation of aqueous sulfur (IV) by nitrogen dioxide, in: Pruppacher H.R. et al. (coordinators) Precipitation scavenging, dry deposition and resuspension. Proceedings of the 4 th International conference, Santa Monica (CA), 29 November3 December 1982, New York, Elsevier, 2v.; v.I, 453-470. Lovelock J.E. (1975) Natural halocarbons in the air and in the sea, «Nature», 256, 193-194. Lunak S., Veprek-Siska J. (1976) Photochemical autooxidation of sodium sulfite catalysed by ferric ions, «Collection of Czechoslovak Chemical Communications», 41, 3495-3503. Maahs H.G. (1983) Kinetics and mechanism of the oxidation of sulfur(IV) by ozone in aqueous solution with particular reference to sulfur dioxide conversion in nonurban tropospheric clouds, «Journal of Geophysical Research», 88, 10721-10731. Martin L.R. (1984) Kinetic studies of sulfite oxidation in aqueous solutions, in: Calvert J.G. (editor) SO2, NO and NO2 oxidation mechanisms. Atmospheric considerations, Boston (MA), Butterworth, 63 -100. Misra P.K. et al. (1985) Scavening rations of acidic pollutants and their use in long-range transport models, «Atmospheric Environment», 19, 1471-1475. NRC (US National Research Council) - Panel on Atmospheric Chemistry (1976) Halocarbons. Effects on stratospheric ozone, Washington (D.C.), National Academy of Sciences. Penkett S.A. et al. (1979) The importance of atmospheric ozone and hydrogen peroxide in oxidizing sulfur dioxide in cloud and rainwater, «Atmospheric Environment», 13, 123-137. Platt U. et al. (1984) Measurement of nitrate radical concentrations in continental air, «Environmental Science and Technology», 18, 365-369. Rogge W.F. et al. (1993) Quantification of urban organic aerosols at a molecular level: identification, abundance and seasonal variation, «Atmospheric Environment. Part A: General Topics», 27A, 1309-1330. Rowland F.S., Molina M.J. (1975) Chlorofluoromethanes in the environment, «Reviews of Geophysic and Space Physics», 13, 1-35. Salisbury G. et al. (2001) Production of peroxy radicals at night via reactions of ozone and the nitrate radical in the 935 ENVIRONMENTAL TECHNOLOGIES marine boundary layer, «Journal of Geophysical Research», 106, 12669-12688. Schwartz S.E. (1984) Gas-aqueous reactions of sulfur and nitrogen oxides in liquid- water clouds, in: Pruppacher H.R. et al. (coordinators) Precipitation scavenging, dry deposition and resuspension. Proceedings of the 4th International conference, Santa Monica (CA), 29 November-3 December 1982, New York, Elsevier, 2v.; v.I, 173-208. Seigneur C. (2001) Current status of air quality models for particulate matter, «Journal of the Air & Waste Management Association», 51, 1508-1521. Seinfeld J.H. (1986) Atmospheric chemistry and physics of air pollution, New York, John Wiley. Weiss R.E. et al. (1977) Sulfate aerosol: its geographical extent in the Midwestern and Southern United States, «Science», 195, 979-981. 936 Wendisch M. et al. (1996) Vertical profiles of aerosol and radiation and the influence of a temperature inversion: measurement and radiative transfer calculations, «Journal of Applied Meteorology», 35, 1703-1715. WMO (World Meteorological Organization)/United Nations Environment Programme (1994) Scientific assessment of ozone depletion, Genève, WMO, Global Ozone Research and Monitoring Project, Report 37. Yung Y.L. et al. (1980) Atmospheric bromine and ozone perturbations in the lower stratosphere, «Journal of the Atmospheric Sciences», 37, 339-353. Ivo Allegrini Consiglio Nazionale delle Ricerche Istituto sull’Inquinamento Atmosferico Monterotondo, Roma, Italy ENCYCLOPAEDIA OF HYDROCARBONS