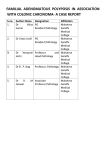
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
1 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) The experience of the Colorectal Cancer Study Group of the University of Modena and Reggio Emilia M. Ponz de Leon, F. Domati, L. Roncucci, G. Rossi, R. Sassatelli, P. Benatti, C. Di Gregorio, M. Pedroni, S. Maffei, D. Nozzi, L.Reggiani Bonetti, E. Borsi, C. De Gaetani, A.Merighi 2 AUTHORS Maurizio Ponz de Leon Professor of Internal Medicine, Dipartimento di Medicine e Specialità Mediche, Università di Modena e Reggio Emilia (Italy) Federica Domati Research Fellow, Dipartimento di Medicine e Specialità Mediche, Università di Modena e Reggio Emilia (Italy) Luca Roncucci Associate Professor of Internal Medicine, Dipartimento di Medicine e Specialità Mediche, Università di Modena e Reggio Emilia (Italy) Giuseppina Rossi Research Fellow, Dipartimento di Medicine e Specialità Mediche, Università di Modena e Reggio Emilia (Italy) Romano Sassatelli Endoscopist, Endoscopy Unit, Reggio Emilia Hospital (Italy) Piero Benatti Aggregate Professor of Internal Medicine, Dipartimento di Medicine e Specialità Mediche, Università di Modena e Reggio Emilia (Italy) Carmela Di Gregorio Pathologist, Pathology Unit, Carpi Hospital (Italy) Monica Pedroni Chief Technician, Dipartimento di Medicine e Specialità Mediche, Università di Modena e Reggio Emilia (Italy) Stefania Maffei Research Fellow, Dipartimento di Medicine e Specialità Mediche, Università di Modena e Reggio Emilia (Italy) Daniela Nozzi Research Fellow, Dipartimento di Medicine e Specialità Mediche, Università di Modena e Reggio Emilia (Italy) Luca Reggiani Bonetti Pathologist, Dipartimento di Patologia Università di Modena e Reggio Emilia (Italy) Enrica Borsi Research Fellow, Dipartimento di Medicine e Specialità Mediche, Università di Modena e Reggio Emilia (Italy) Carmela De Gaetani Pathologist, Dipartimento di Patologia Università di Modena e Reggio Emilia (Italy) Alberto Merighi Endoscopist, Dipartimento di Medicine e Specialità Mediche, Università di Modena e Reggio Emilia (Italy) Umana, Umana, 3 Addresses of the Study Group: • Struttura Complessa di Medicina 1, Dipartimento di Medicine e Specitalità Mediche, Policlinico, Via del Pozzo 71, 41100 Modena (Italy) • Servizio di Anatomia Patologica, Ospedale di Carpi, Via G. Molinari 2, 41012 Carpi (Modena) • Struttura Complessa di Anatomia Patologica, Dipartimento di Servizi Diagnostici, di Laboratorio e di Medicina Legale, Policlinico, Via del Pozzo 71, 41100 Modena Telephones: M.Ponz de Leon +39 059.4222269 Secretary (G.Rossi) +39 059.4224715 P.Benatti +39 059.4225895 L.Roncucci +39 059.4224052 C.Di Gregorio +39 059.659480 C.De Gaetani +39 059.4224816 Fax: Secretary (MO) +39 059.4222958 Anatomia Patologica (MO) +39 059-4224820 Anatomia Patologica Carpi +39 059-659469 E-mail: M.Ponz de Leon [email protected] ([email protected]) P.Benatti [email protected] L.Roncucci [email protected] C.Di Gregorio [email protected] C.De Gaetani [email protected] Website: www.tumoricolorettali.unimore.it 4 CONTENTS PRESENTATION Page 6 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) Page 7 Introduction and Definition Page 9 Diagnosis, Clinical Features, Morphology Page 10 Extracolonic Manifestations Page 12 Molecular Biology and Genotype-Phenotype Correlations Page 13 Surveillance and Treatment: Surgery versus Endoscopy Page 17 Genetic Counseling Page 18 Conclusions Page 23 References Page 25 DESCRIPTION OF FAMILIES WITH ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS OBSERVED IN MODENA BETWEEN 1984 AND 2009 Page 36 SUMMARY TABLES OF THE MAIN RESULTS OBSERVED IN THE 27 FAMILIES WITH AFAP Page 97 5 PRESENTATION After the volumes on Lynch syndrome (2007) and Familial Adenomatous Polyposis (2008), in 2009 the Colorectal Cancer Study Group of the University of Modena and Reggio Emilia presents a volume which summarizes the experience with Attenuated Familial Adenomatous Polyposis (AFAP). The senior members of the Group initiated to study Hereditary Colorectal Neoplasms in 1983, when families with clinical features of Familial Adenomatous Polyposis (FAP) and Lynch syndrome were observed and characterized. While Lynch syndrome was virtually unknown, FAP had already been described by almost 100 years; yet the syndrome was still poorly understood in Italy, and clinical experience limited to a few centers. The disease had been studied mainly in the United Kingdom (where a Polyposis Registry was instituted since 1925, at St. Mark’s Hospital, London) and in the United States, but in many other European countries the knowledge on FAP was as limited as in Italy. In 1985, experts of several countries constituted the “Leeds Castle” Polyposis Group (from the place of the first meeting, in Kent, U.K.) with the purpose of promoting the research on colorectal polyposis (FAP and related syndromes). And this is what happened, since in those years (1985-1995) there were several advancements, including the definition of extracolonic manifestations, the development of chemoprevention and, above all, the identification of APC gene. In 1998, the Leeds Castle Group joined with the International Study Group on Lynch syndrome, giving origin to the InSiGHT, the Association which gathers all researchers interested in familiality and clinical genetics of tumors of the digestive organs. The Colorectal Cancer Study Group of the University of Modena and Reggio Emilia played an active role in this long period of rewarding and fruitful scientific activity. Moreover, in the years Ninety of the XX century the Group was among the promoters of the constitution of AIFEG (Associazione Italiana per lo studio della Familiarità ed Ereditarietà Gastrointestinale), the Society which represents Italian researchers interested in the study of Polyposis and Lynch syndrome. 6 Along these years, the Study Group continued in the scientific way traced at the beginning of its activity, that is, identification, management, biological and molecular characterization of Hereditary Colorectal Cancer Syndromes. The institution, in 1984, of a specialized colorectal cancer Registry, in the Health Care District of Modena, was of great help in reaching these objectives. At present, 64 families with clinical features of FAP have been identified (41 of which with constitutional mutations in either APC or MutYH genes), 34 families with Lynch syndrome (all with germline mutations in either MSH2, MLH1 or MSH6 genes), 32 families with clinical HNPCC (i.e., clinical features of Lynch syndrome but without documented constitutional mutations), and 6 families with clinical features suggestive of Peutz-Jeghers syndrome. The first part of this volume deals with general aspects of AFAP, such as definition, molecular biology, clinical features and management. In the second part the individual families identified between 1984 and 2009 are illustrated in details, with a schematic genealogical tree, a list of the main clinical data of affected patients, a concise family history and the results of biomolecular tests. In the last part of the volume summary tables of the main data are presented. The objective of the volume is two-fold. First, to illustrate our group of patients and families, pointing out the main clinical and biological challenges related to the diagnosis of AFAP. Second, to make accessible to physicians – both specialists and family doctors – a topic which remains poorly defined and difficult to manage, especially for its genetic basis and the consequent implications for family members of the proband. In 2005 a screening campaign against colorectal tumors was initiated in Region Emilia-Romagna. One of the effects of this Public Health intervention was to alert physicians (and the general population) on the clinical, epidemiological and social relevance of colorectal neoplasms. It seems to us appropriate, with the present volume, to illustrate a rare but particularly interesting clinical entity which represents an ideal model for the study of colorectal tumorigenesis. 7 Finally, we would like to express our gratitude to all individuals – doctors and patients – who offered their help in the study of Attenuated Polyposis and, thus, in the collection and analysis of the data reported in this volume. In particular, we would like to thank: Dr.ssa Alessandra Viel, Centro di Riferimento Oncologico, Aviano, Pordenone Prof. Maurizio Genuardi, Genetica Medica, Università di Firenze Dr.ssa Tiziana Venesio, Centro Oncologico, Candiolo, Torino Dr.ssa Liliana Varesco, Istituto Nazionale per lo Studio dei Tumori, Genova Prof. Francesco Tonelli, Chirurgia Generale, Università di Firenze Prof. Giovanni Lanza, Anatomia Patologica, Università di Ferrara and their collaborators, with whom we shared at least 20 years of continuous and enthusiastic investigation, and we continue to collaborate. The Authors 8 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) INTRODUCTION AND DEFINITION Familial Adenomatous Polyposis (FAP) is characterized by the presence of at least 100 adenomatous lesions of the large bowel, several extracolonic manifestations and an autosomal dominant type of genetic transmission (1, 2). The disease is caused by constitutional mutations in the APC gene (for Adenomatous Polyposis Coli) (3, 4), and, to a lesser extent, by mutations of the recently identified MutYH gene (5, 6). At variance with classic FAP, Attenuated Familial Adenomatous Polyposis (AFAP) is characterized by the presence of < 100 synchronous adenomas of the large bowel (7, 8); extracolonic manifestations are not so frequent as in FAP (9), and in most cases only one individual is affected in a given family (10); moreover, the relative contribution of APC and MutYH to the AFAP phenotype has to be defined (11). As a matter of fact, the term “attenuated” refers not only to the number of adenomas, but also to the milder clinical course of the disease, featured by a later onset of colorectal adenomas and carcinoma, and a limited expression of extracolonic changes. Lynch et al (12) and Leppert et al (13) were presumably the first to draw attention to this new phenotype, by describing families with several adenomas and often malignancies of the large bowel but not meeting the clinical criteria for FAP. Since then, several other families with this attenuated form of polyposis have been reported; however, AFAP still remains a poorly defined disease, despite the attempt to suggest precise diagnostic criteria (14). Probably the simplest way to define the syndrome is to consider as AFAP any case presenting with a number of synchronous adenomatous lesions of the large bowel ranging between 10 to 99, unregarding the age of onset, the presence of constitutional mutations, extracolonic changes and features of autosomal dominant or recessive transmission. If this definition has the great advantage of simplicity, it should be acknowledged that in some cases (i.e., patients with 10 to 15 adenomas) the differential diagnosis from multiple polyps of the large bowel may be difficult, if not impossible. By the same token, in presence of 90 9 to 99 adenomatous lesions the distinction between AFAP and FAP might reveal rather hard. Molecular biology does not seem to be of help, since at variance with FAP (where constitutional mutations are usually detected in 80 to 95% of families) germline alterations can be identified in 30-50% of patients and families with attenuated polyposis (15). Since the definition is unclear and no real consensus exists, incidence and frequency of AFAP are difficult to establish. In the Dutch experience less than 10% of FAP patients showed the attenuated phenotype (16), whereas in the large series of Friedl et al AFAP was diagnosed in approximately 15% of FAP families (17), and even lower estimates were reported by other studies (18). However, it is worth noting that in none of these investigations a systematic search for Attenuated Polyposis – through endoscopic records or data of a cancer Registry – was carried out or attempted. At present AFAP is recognized as a distinct clinical entity, based on the presence of attenuated clinical features as compared with FAP. Whether such distinction between FAP and AFAP is warranted remains difficult to anticipate, since future biomolecular investigations might reveal that the syndrome at present defined as AFAP is only part of a larger entity based on APC and MutYH constitutional alterations. Moreover, the possible involvement of other genes implicated in the mechanisms of DNA base excision repair in inducing the AFAP phenotype cannot be excluded (19). DIAGNOSIS, CLINICAL FEATURES, MORPHOLOGY Because of uncertainties in defining the syndrome, not all series which appeared in the literature respected the upper limit of 90-100 adenomas in the large bowel, and the lower limit of 10 lesions was even less taken into account. For instance, in a large series published in 1998 (20), the mean number of adenomas was 44, but the range between 1 and 500. The Dutch Polyposis Registry recently defined AFAP as follows: 1) at least two first-degree relatives with 10-99 colorectal adenomas diagnosed after the age of 30 years; 2) one patient with 10-99 adenomas diagnosed after the age of 30 years plus a first-degree relative with 10 colorectal cancer and a few (< 10) adenomas; 3) no family members with “classic” FAP (> 100 adenomas) before the age of 30. Applying this definition, 25 out of 315 FAP families (8.0%) met the clinical criteria for attenuated polyposis (21). According to some authors, the majority of patients with AFAP show a prevalent proximal distribution of adenomas (i.e., from the cecum to the splenic flexure), and these are often of the flat type (22, 23). However, this does not seem to be a general rule, and in many patients the lesions are distributed exclusively, or predominantly, in the left colon, as it occurs in classic FAP (24). Some studies reported a tendency to spearing the rectum from adenomas in AFAP individuals (25). As far as the age of onset of adenomas is concerned, this is usually delayed in AFAP when compared with the pattern observed in FAP. Thus, Scott et al reported that individuals with Attenuated Polyposis were diagnosed, on average, more than 15 years later than FAP patients (15). Among symptoms leading to diagnosis, rectal bleeding and abdominal discomfort are by far the most frequent (26). Owing to the small size of many AFAP families, and the prevalence of solitary cases, diagnosis for screening is less frequent than in FAP (27). In most series, the mean age of adenoma development ranges between 40 and 50 years (21, 28), and in approximately half of the patients cancer is already present at diagnosis. In most series, Attenuated Adenomatosis Coli affects the two sex at the same extent, a feature which is consistent with autosomal transmission; however, there are reports of a striking gender effect (with female preponderance) in AFAP patients (28, 29, 30). Histologically, colorectal adenomas which develop in AFAP do not seem to be different from sporadic lesions as well as from polyps observed in profuse polyposis. Tubular or tubulovillous adenomas with low, medium or severe dysplasia (low and high grade) are the most frequent pathological findings in these patients. However, according to some authors flat lesions seem particularly frequent in AFAP (22, 23, 31), and this may complicate diagnosis and follow-up (32). In addition, Boporai et al (33) recently reported the frequent occurrence of hyperplastic polyps and of sessile serrated adenomas especially in the subgroup of individuals with constitutional mutations in the MutYH gene, thus suggesting that these types of polyps could be associated causally with deficiency of 11 MutYH function and, in turn, that different morphological pathways might exist between APC-gene and MutYH-gene related polyposis. EXTRACOLONIC MANIFESTATIONS In classic FAP extracolonic manifestations are usually observed in almost all families, and in some cases they can alert the physician on the existence of the syndrome. In Attenuated Polyposis there are several reports of extracolonic changes, however, their frequency seems lower than in FAP; what remains unclear is whether this low incidence reflects a biological feature of AFAP, or should be attributed to less attention in searching or reporting alterations which are not limited to the large bowel. Thus, there is very limited information on the frequency of Congenital Hypertrophy of the Retinal Pigmented Epithelium (CHRPE) in patients with Attenuated Familial Polyposis (34); similarly, other common features of FAP such as osteomas, supernumerary teeth, epidermoid cysts, hepatoblastoma and thyroid carcinoma have been reported only occasionally. The frequency of desmoid tumors is in the order of 10-15% of all patients (and families) with FAP, but their incidence in AFAP has not been determined, though several authors reported an increased risk of desmoids in some families with the attenuated phenotype and specific APC constitutional mutations (35, 36). There are also reports of families with a marked inheritance of desmoid tumors (“Hereditary Desmoid Diseases”, according to some authors) associated with clinical features of Attenuated Polyposis (37, 38). Glandular polyps of the stomach and duodenal adenomas are undoubtedly the most frequent extracolonic manifestations observed in FAP as well as in AFAP. However, if there is a general consensus on a reported frequency of gastric lesions in about 50% (though much higher in Japanese series) (39, 40), and of duodenal adenomas in 90-100% of patients with classic FAP, the reported frequency of these lesions appears to be lower in AFAP, and with conflicting results. Thus, in one series gastroduodenal lesions (mostly glandular polyps of the fundus and antrum) were found in nearly 70% of patients with Attenuated Polyposis 12 (41), while other authors reported much lower rates (42, 43). Upper gastroduodenal malignancies (periampullary lesions in most cases) have been detected in only few patients with AFAP (44, 45). MOLECULAR BIOLOGY AND GENOTYPE-PHENOTYPE CORRELATIONS The AFAP phenotype can be associated with constitutional mutations of at least two genes: APC and MutYH. This means that in some cases the disease is transmitted through an autosomal dominant model (APC), while in other cases through a recessive type of inheritance. How many cases of AFAP should be attributed to APC alterations and how many to MutYH changes is still a matter of concern, although both genes play an important role, and the prevalent attitude is to consider two forms of polyposis, one associated with APC (APC-related FAP) and one with MutYH (MutYH related FAP, more simply, MAP) (46). It is likely, however, that germline alterations of other genes contribute to the AFAP phenotype, including genes of base-excision repair and those of DNA mismatch repair machinery (47, 48). Finally, owing to the frequency of adenomatous polyps in the general population and to the lack of stringent criteria for defining AFAP, it is also possible that in a given fraction of all cases the disease might be due to environmental factors, presumably inducing somatic DNA alterations in genes crucial for cell replication and differentiation (49, 50). APC Related AFAP The APC gene is located on chromosome 5q21, and is constituted by 2,843 codons in 15 exons. APC functions as a tumor suppressor gene and encodes for a 300 KDa protein which interacts with β-catenin. In absence of APC (usually as a consequence of truncating mutations), the binding to β-catenin does not occur, β-catenin accumulates in the cytoplasm and binds to specific transcription factors, thus altering the expression of various genes influencing cell proliferation, differentiation and apoptosis (51, 52). This proliferative response may represent the first step toward cancer development. 13 In individuals with the AFAP phenotype several mutations have been identified in the APC locus. It is commonly assumed that the disease is due to mutations in the extreme portions of the gene, 5’ or 3’ ends. However, from the available literature it seems that these alterations are not clustered in a specific DNA segment of the gene, but encompass exons 3, 4, 5, 6, 9 and a large fraction of exon 15 (approximately from codon 1000 to 3000) (53, 54). No clear and well established genotype-phenotype correlations have been revealed. Thus, number of polyps in the large bowel, presence of extracolonic manifestations and morphologic features of adenomas in APC-associated AFAP do not seem to be related to specific germline alterations. This is not surprising, since in FAP patients correlations between type of mutation and expressed phenotype could be established only in large groups of individuals, while the phenotype cannot be predicted in individual subjects. In addition, several observations showed a marked phenotypic variability in patients carrying the same APC mutation (55, 56). The prevalence of APC mutations among individuals with clinical features of Attenuated Polyposis remains poorly defined. In a recent study, Nielsen et al (21) reported that approximately 1/3 of their AFAP patients were positive for APC mutations; however, these families were selected on the basis of stringent clinical criteria, concerning not only the number of colorectal adenomas, but also age at diagnosis and the presence of affected first-degree relatives. With a different and less stringent definition of AFAP, a lower prevalence of mutations should be expected. MutYH Related AFAP The MutYH gene (previously known as MYH) encodes for a glycosylase involved in the base excision repair caused by oxidative damage to DNA (57); the gene, located at chromosome 1p32-34, is deleted in several human malignancies (58). In 2002, Al Tassan et al reported the presence of biallelic MutYH mutations in a family with several members affected by multiple colorectal adenomas and carcinoma (5). Further investigations revealed that homozygous or compound heterozygous mutations of MutYH could be detected in 20 to 30% of AFAP patients and in approximately 10% of classic FAP cases (59, 60). Thus, 14 after nearly 20 years from the discovery of APC, a second gene specifically associated with the adenomatosis phenotype was identified; moreover, the studies showed an unsuspected role of the base excision repair system in genetically determined colorectal neoplasms. In Western countries, the two missense mutations Y165C and G382D (Y179C and G396D according to other authors) account for the large majority of disease-causing alterations (61), while in some Eastern populations the nonsense E466X mutation seems to be the most frequent (62). In accordance with an autosomal recessive type of genetic transmission, biallelic mutations are required for the development of polyposis or cancer; indeed, no unaffected carrier of biallelic MutYH mutations has been identified, thus suggesting a high penetrance of the trait (63). It is still undefined whether the heterozygous condition carries an increased risk of adenomas or carcinoma in the large bowel or other organs. Immunohystochemical studies indicated that neoplasms developed in MutYHassociated polyposis show disappearance of staining from the nucleus, and segregation of immunoreactivity in the cytoplasm, thus suggesting a possible role of this technique in the screening for individuals at risk of MAP (64). Other authors, however, were unable to find out distinctive pathological features of the immunohistochemical pattern which could predict the presence of MutYH mutations in colorectal cancer (31). Recent studies suggested interesting genotype-phenotype correlations in MAP. Thus, Nielsen et al (65) showed that patients with biallelic G396D (or compound G396D/Y179C) mutations presented later with clinical features of polyposis and had a lower risk of developing colorectal cancer than patients with homozygous Y179C mutations. It seems therefore that some alterations can be associated with a more severe phenotype and clinical course, while other mutations with a relatively milder disease. At variance with APC, whose role in AFAP is not precisely defined, several studies indicated that MutYH is mutated in 20 to 40% of all patients and families with clinical features of Attenuated Polyposis (30, 66, 67). Thus, in families with suspicion of recessive inheritance (i.e., lack of vertical transmission of the trait cancer or polyposis) or when neoplasms show evidence of DNA excision repair damage – such as G T transversions 15 at codon 12 of the K-ras gene (68) – molecular studied should begin with a detailed sequence analysis of the MutYH gene. SURVEILLANCE AND TREATMENT: SURGERY VERSUS ENDOSCOPY At variance with FAP – in which surveillance and treatment follow precise guidelines that have been improved over time (69) – no consensus exists on the management of Attenuated Polyposis and, consequently, the clinical approach to the disease remains largely empirical. Mutation analysis of APC and MutYH genes assumed considerable importance in the most recent years, not only for being of help in differentiating between FAP and AFAP, but also for excluding the presence of other inherited cancer syndromes – such as Lynch syndrome (70) or its variant Muir-Torre syndrome (71) – that may in some cases mimic AFAP. For subjects who test positive for APC or MutYH (biallelic) mutations associated with clinical features of AFAP, baseline colonoscopy together with esophagogastroduodenoscopy should be carried out at the time of genetic testing or by the age of 14-16 years; in case of negative results, the investigation should be repeated after 2-3 years and then at regular intervals of time, since late appearance of adenomas is frequent (72). It should be noted however that in most series some 30 to 50% of patients may result negative for constitutional mutations and, consequently, management should be based only on clinical findings (73). The main clinical decision to be taken in patients with Attenuated Polyposis is the choice between endoscopic or surgical approach to the disease. In absence of well defined guidelines, a purely empirical approach could be traced as follows. Patients with clinical features of AFAP, but with a number of polyps which can be removed through endoscopy should undergo polypectomy with continued yearly surveillance. It is important to analyze all removed lesions, since the presence of severe dysplasia in one or more adenomas might change the conservative approach into a more radical treatment. Patients with adenomatous 16 lesions too numerous to be removed endoscopically, or for whom endoscopic surveillance is not possible (low compliance), should be considered for surgical management (7, 74). Patients with Attenuated Polyposis are at increased risk of developing colorectal cancer when compared with the general population; however, the exact risk is difficult to calculate, and from the available observations does not seem as “absolute” (that is, nearly 100%) as for FAP patients. It follows that indications for prophylactic colectomy differ between the two conditions. First of all, many AFAP patients are treated lifelong with the sole endoscopic approach, especially when up to 20 polyps are present in the large bowel and no severe dysplasia is observed at histology (74, 75). Second, when the excessive number of polyps hampers the complete endoscopic removal but the rectum is spared from lesions (or it shows a few small lesions that can easily be removed), subtotal colectomy and ileorectal (or ileosigmoid) anastomosis are usually carried out, instead of the technically more difficult proctocolectomy with ileoanal anastomosis and J pouch (76). In case of this choice, it is quite obvious that the rectal stump should require continuous surveillance, owing to the high risk of newly developed lesions (77). Finally, when multiple adenomas are clustered in a single segment of the large bowel – especially in the proximal intestine – segmental resection or hemicolectomy may be valuable options (78), providing of course that the patient is willing to undergo close endoscopic surveillance of the remaining large bowel. Treatment and surveillance can also be influenced by recent advancements in molecular biology. In a collaborative investigation, Bulow et al. (76) recommended to execute colectomy and ileorectal anastomosis (but not ileoanal anastomosis) in FAP or AFAP patients with APC constitutional mutations at codon 0-200 or after codon 1500, at the two extremes of the gene. In contrast, according to some authors, mutations in the central zone of the gene (around codon 1309), usually associated with a severe phenotype and profuse polyposis, would benefit of proctocolectomy and ileoanal anastomosis (79, 80). In a recent study, Nielsen et al identified biallelic MutYH mutations associated with a severe phenotype (Y179C), thus suggesting that even for MutYH gene alterations surveillance and management (of AFAP patients) might be somehow guided by the molecular findings (65). 17 Other investigations suggested that certain drugs – especially nonsteroideal antiinflammatory compounds – may play a role in reducing number or dimensions of adenomas, especially in classic FAP (81, 82). It should be noted, however, that most studies showed incomplete polyp regression, and over short follow-up periods (83). Sulindac, Celecoxib and Rofecoxib (the second and third COX-2 inhibitors) were the most effective in reducing the polyp burden. There is little or no information on the possible role of these drugs in Attenuated Polyposis. Two major factors, however, should be taken into account. First, there is evidence of colorectal cancer occurrence during attempts to induce polyp regression through the use of anti-inflammatory compounds (84). Second, recent observations reported an increased cardiovascular risk and thrombotic effects among patients treated with COX-2 inhibitors in adenoma chemoprevention trials (85), thus raising some concerns about the appropriate use of these new drugs. Treatment of upper gastrointestinal lesions is difficult in classic FAP and may be complex also in Attenuated Polyposis, at least in individual cases (86). Most physicians recommend gastroduodenoscopy at regular intervals of time for AFAP patients, and gastric polyps (usually glandular or hyperplastic lesions) or duodenal adenomas may precede, in some patients, the appearance of colorectal tumors (87). As in the case of FAP, optimal treatment and surveillance of upper gastrointestinal lesions, and the possible advantage of chemoprevention (with nonsteroideal anti-inflammatory drugs, Ursodeoxycholic acid or other chemical compounds) remain still undefined (88). GENETIC COUNSELING In general terms, with genetic counseling the physician tries to acquire all relevant information concerning a given individual, his (or her) family, and the disease – presumably genetic – object of counseling, In a successive phase the same physician (a geneticist, a gastroenterologist, an internist or the family doctor) suggests a given strategy, which may imply the execution of genetic tests or other clinical examinations, changes of diet and lifestyle, as well as medical or surgical interventions. 18 In Attenuated Familial Adenomatous Polyposis genetic counseling follows in general terms the same guidelines adopted from many years for classic FAP; however, while in FAP germline mutations can be detected in 80-90% of patients with the typical phenotype (89), in AFAP the fraction of positive families is in the order of 50% or less, at least in most series (21). Moreover, many families present as single cases, especially when constitutional mutations are not detectable, while in classic FAP segregation of the deleterious tract can be followed in most families through various generations (90). As already discussed, two genes – APC and MutYH – have been associated with the AFAP phenotype, but with different types of genetic transmission. Genetic counseling in APC-associated AFAP In APC-related AFAP genetic counseling starts with a detailed description of the pedigree, usually through the proband. In most AFAP cases the nuclear pedigree will contain all relevant information; only few cases usually require the extension of the family tree to second and third degree relatives (as in classic FAP). When possible, cases of cancer or polyposis should be verified by clinical charts, histological reports or other certificates. In tracing genealogical trees, conventional symbols are used to identify the proband (arrow), the sex (square for man, circle for woman), death of the patient (diagonal bar), generations (latin numbers), and single components of a sibship (arabic numbers); horizontal and vertical lines join husband and wife, siblings and offsprings. There are not conventional symbols for indicating the diseases. The symbols used in the present volume are indicated inside the cover. In the clinical suspicion of AFAP, genetic testing (i.e., search for constitutional APC mutations) should initially be proposed to the proband; if a deleterious mutation is detected, the test is offered, as an option, to other family members at risk identified through the pedigree. It should be noted, and emphasized, that the decision to undergo genetic testing is strictly personal, should be discussed with the patient and requires a written formal consent (91). It is entirely possible, and should not surprise the physician, that a certain number of 19 high-risk individuals show indifference or even suspicion towards genetic testing; these subjects – who might be defined “fatalistic” – probably believe, or suppose, that to became aware of a given predisposition towards diseases (or a specific disease) might alter their psychological equilibrium, which sometimes is reached and maintained with difficulty. In these cases the genetic counselor should not exert any pressure, but just limit his (her) contribution to a correct information. In gene carriers within typical FAP families the first colonoscopy is usually suggested at puberty, when adenomas begin to appear in most patients. Presumably the same recommendation holds true for AFAP, in which however a later appearance of polyps can be expected. Within a few years gastroduodenoscopy should be executed, in order to find out gastric or duodenal lesions. In solitary cases (i.e., only one affected member), the proband’s offsprings carry a 50% risk to be affected (in accordance with the autosomal dominant type of transmission), and genetic testing should be proposed. When in a given family no APC mutations are identified, then genetic testing is unable to detect constitutional alterations despite the presence of a phenotype consistent with AFAP. This might be due to several factors, including: a) procedural errors (which are always possible), b) involvement of other genes, especially MutYH, c) truly sporadic cases of AFAP, d) multiple adenomas of the large bowel which mimic AFAP. When no APC (or MutYH) mutations are detected in a given family or individual, the genetic test loses any predictive value, and cannot identify cases at risk of polyposis. In these subjects – who probably represent the larger fraction of AFAP – first-degree relatives of the proband should be followed-up only on a clinical basis, starting with endoscopic surveillance at puberty and continuing lifelong at regular intervals of time (92, 93). Genetic counseling in MutYH-associated Polyposis (MAP) According to some series, nearly one third of all Attenuated Polyposis can be attributed to biallelic constitutional mutations in the MutYH gene (59, 60). At variance with APC, MutYH recognizes an autosomal recessive type of transmission, thus, clinical 20 manifestations appears only in case of biallelic (homozygous or compound heterozygous) mutations. In most instances, affected individuals have healthy parents, though heterozygous carriers of the mutation, and similarly unaffected will result the offsprings. The pattern of transmission is markedly different from that of APC-related polyposis: no “verticality”, but presence of affected individuals in a single generation or sibship. By extending the genealogical tree, other affected individuals can be identified, but with intervals of unaffected generations. As in all recessive conditions, the transmission of MutYH mutations is difficult to recognize when compared with APC-positive families, either for the “horizontal” pattern of transmission, or for the small size of most modern families. Since the risk that an individual with heterozygous parents results affected is in the order of 25% (1 in 4), it follows that in the majority of cases AFAP associated with MutYH mutations will appear as sporadic, without any family history suggestive of a genetic defect. Main recommendations concerning diet and style of life Carriers of constitutional mutations of the APC or MutYH genes are at increased risk of developing adenomatosis and cancer of the large bowel. The risk approaches 100% for FAP patients, while for Attenuated Polyposis it is still undefined, though much higher than that of the general population (94). A similar risk can be expected for AFAP individuals in whom germline alterations cannot be detected. Progression from normal appearing mucosa to adenomas and cancer depends on several factors, including, presumably, the type of mutation, the role of modifier genes (95), and the possible interaction between genotype and several environmental agents (96). As already discussed, the main recommendations for gene carriers (and, more in general, for individuals at risk) concern screening and endoscopic surveillance of the large bowel, and, after surgery, the rectal stump and ileal reservoir; moreover, a careful follow-up of the upper gastrointestinal tract is also suggested. The ongoing clinical trials should provide an answer to the possible role of chemoprevention, especially with Cox-2 inhibitors (97). 21 At present there is no evidence that diet and style of life can be of help in influencing the clinical course of patients with FAP or AFAP phenotypes. However, there is some consensus on the relevance of some protective or risk factors in the pathogenesis of colorectal neoplasms, despite the poor reproducibility of several observations (98, 99). Although most of the studies refer to sporadic tumors, rather intuitively the same implications might be relevant also for individuals with inherited colorectal neoplasia, such as Lynch syndrome, FAP or Attenuated Polyposis. Following this line of reasoning, it is likely that at least the following suggestions on diet and style of life might be given to patients with FAP or AFAP: 1. It is important to avoid a fully sedentary life, especially for those individuals whose job activity does not imply physical exercise (the large majority of the population, at present); 2. Similarly relevant is to reach and maintain an ideal body weight, avoiding overweight and obesity; 3. We would suggest to quit cigarette smoking, and to limit the ingestion of alcoholic beverages (100, 101); 4. As far as the diet is concerned, the situation is even more complex (102, 103). However, the main suggestions remain the following: - to enrich the diet of dry fiber, such as bran; - to increase the consumption of fruit and vegetables; - to limit the ingestion of meat and animal fat. 22 CONCLUSIONS Attenuated Familial Adenomatous Polyposis is a relatively new clinical entity described approximately 20 years ago (12, 13) and since then actively investigated. However, AFAP remains poorly defined, and there are still concerns whether or not it represents a disease, or a syndrome, entirely separated from FAP, on one extreme, and from Multiple Adenomas of the large bowel on the other extreme. AFAP is characterized by a milder phenotype when compared with classic FAP, not only for the absolute number of adenomas, but also regarding the age of onset of colorectal neoplasms (both benign and malignant) and the frequency of extracolonic manifestations. Moreover, although the same genes (APC and MutYH) seem to be involved, some 30 to 50% of AFAP patients, or even more, remain without a genetic diagnosis (59, 60). The main difficulty with AFAP is a proper definition of the disease, since clinical criteria have not been defined, and are difficult to define. If we apply strict clinical criteria, taking into account not only the number of adenomas, but also the age of onset of polyps and familial aggregation of neoplasms, then we could miss many cases. On the other hand, by accepting a simple definition of AFAP – such as the presence of less than 100 adenomas in the large bowel, unregarding age of onset and family history – it is likely that some patients with sporadic multiple polyps (104) could be included among AFAP cases. In fact, to distinguish AFAP from the group of multiple adenoma patients is extremely difficult on a clinical ground, owing to the similar phenotypes, and the possible presence of a family history of polyps and cancer in both categories of patients. It should be noted, however, that the diagnosis of FAP is based only on the phenotype (> 100 synchronous adenomas of the large bowel), and does not take into account the age of onset of the lesions or the presence of “verticality” (105). By analogy, therefore, the definition of AFAP might be based mainly, if not exclusively, on the phenotype of the proband. In the present volume we selected families on the basis of a simple definition of AFAP: the presence in the large bowel of a number of synchronous adenomas in the range 10 to 99. We considered this definition as the most appropriate, especially in analogy with the accepted clinical definition of FAP. This does not mean that age of onset of adenomas 23 (or cancer), dominant or recessive type of transmission, and presence of constitutional mutations are not important; however, some of these parameters could be viewed as criteria of exclusion for many AFAP patients. Again, by analogy with the well defined classic Polyposis, up to 30% of typical FAP patients appear as “solitary cases”, and this means without a family history, often with a late age of onset, and in some cases without a molecular diagnosis (106). Unavoidably, the choice to adopt a rather “loose” definition of AFAP renders the clinical condition extremely heterogeneous. It is likely therefore that AFAP patients described in the present volume reflect a spectrum of diseases which spans from purely genetic cases of FAP and other Hereditary Cancer syndromes to sporadic multiple adenomas of the large bowel. The other relevant and unsettled aspect of AFAP is treatment. On the basis of clinical findings, we should expect a better prognosis in AFAP compared to classic FAP; however, evidence of this contention if lacking, and, consequently, several uncertainties remain on the choice of the optimal approach. Owing to the high risk of cancer, and to the possibility of overlooking neoplasms during surveillance (mainly for the poor compliance of many patients) many authors favor prophylactic colectomy, usually with ileorectal anastomosis, and consider surgery as the optimal therapy (76). However, an increasing number of AFAP patients are managed through endoscopy, especially when the polyps are not too numerous. More observations and, possibly, controlled clinical trials are required to define standard guidelines of treatment in Attenuated Polyposis. 24 REFERENCES 1. Lockhart-Mummery P. Cancer and heredity. Lancet 1925;1:427-9. 2. Alm T, Dencker H, Lunderquist A, Mark J, Mitelman F, Norryd C, Tranberg KG. Gardner's syndrome. Acta Chir Scand. 1973;139(7):660-5. 3. Bodmer WF, Bailey CJ, Bodmer J, Bussey HJ, Ellis A, Gorman P, Lucibello FC, Murday VA, Rider SH, Scambler P, et al. Localization of the gene for familial adenomatous polyposis on chromosome 5. Nature. 1987 Aug 13- 19;328(6131):614-6. 4. Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB, Smith KJ, Preisinger AC, Hedge P, McKechnie D, et al. Identification of FAP locus genes from chromosome 5q21. Science. 1991 Aug 9;253(5020):661-5. 5. Al-Tassan N, Chmiel NH, Maynard J, Fleming N, Livingston AL, Williams GT, Hodges AK, Davies DR, David SS, Sampson JR, Cheadle JP. Inherited variants of MYH associated with somatic G:C-->T:A mutations in colorectal tumors. Nat Genet. 2002 Feb;30(2):227-32. 6. He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998 Sep 4;281(5382):1509-12. 7. Hernegger GS, Moore HG, Guillem JG. Attenuated familial adenomatous polyposis: an evolving and poorly understood entity. Dis Colon Rectum. 2002 Jan;45(1):127-34; discussion 134-6. 8. Galiatsatos P, Foulkes WD. Familial adenomatous polyposis. Am J Gastroenterol. 2006 Feb;101(2):385-98. 9. Knudsen AL, Bisgaard ML, Bülow S. Attenuated familial adenomatous polyposis (AFAP). A review of the literature. Fam Cancer. 2003;2(1):43-55. 10. Burt RW, Leppert MF, Slattery ML, Samowitz WS, Spirio LN, Kerber RA, Kuwada SK, Neklason DW, Disario JA, Lyon E, Hughes JP, Chey WY, White RL. Genetic testing and phenotype in a large kindred with attenuated familial adenomatous polyposis. Gastroenterology. 2004 Aug;127(2):444-51. 11. Marra G, Jiricny J. Multiple colorectal adenomas--is their number up? N Engl J 25 Med. 2003 Feb 27;348(9):845-7. 12. Lynch HT, Smyrk TC, Lanspa SJ, Lynch PM, Watson P, Strayhorn PC, Bronson EK, Lynch JF, Priluck IA, Appelman HD. Phenotypic variation in colorectal adenoma/cancer expression in two families. Hereditary flat adenoma syndrome. Cancer. 1990 Sep 1;66(5):909-15. 13. Leppert M, Burt R, Hughes JP, Samowitz W, Nakamura Y, Woodward S, Gardner E, Lalouel JM, White R. Genetic analysis of an inherited predisposition to colon cancer in a family with a variable number of adenomatous polyps. N Engl J Med. 1990 Mar 29;322(13):904-8. 14. Attenuated adenomatous polyposis coli: the role of ascertainment bias through failure to dye-spray at colonoscopy. Wallace MH, Frayling IM, Clark SK, Neale K, Phillips RK. Dis Colon Rectum. 1999 Aug;42(8):1078-80. 15. Scott RJ, Meldrum C, Crooks R, Spigelman AD, Kirk J, Tucker K, Koorey D; Hunter Family Cancer Service. Familial adenomatous polyposis: more evidence for disease diversity and genetic heterogeneity. Gut. 2001 Apr;48(4):508-14. 16. Nielsen M, Franken PF, Reinards TH, Weiss MM, Wagner A, van der Klift H, Kloosterman S, Houwing-Duistermaat JJ, Aalfs CM, Ausems MG, BröckerVriends AH, Gomez Garcia EB, Hoogerbrugge N, Menko FH, Sijmons RH, Verhoef S, Kuipers EJ, Morreau H, Breuning MH, Tops CM, Wijnen JT, Vasen HF, Fodde R, Hes FJ. Multiplicity in polyp count and extracolonic manifestations in 40 Dutch patients with MYH associated polyposis coli (MAP). J Med Genet. 2005 Sep;42(9):e54. 17. Friedl W, Caspari R, Sengteller M, Uhlhaas S, Lamberti C, Jungck M, Kadmon M, Wolf M, Fahnenstich J, Gebert J, Möslein G, Mangold E, Propping P. Can APC mutation analysis contribute to therapeutic decisions in familial adenomatous polyposis? Experience from 680 FAP families. Gut. 2001 Apr;48(4):515-21. 18. Vasen HF. Clinical diagnosis and management of hereditary colorectal cancer syndromes. J Clin Oncol. 2000 Nov 1;18(21 Suppl):81S-92S. 19. Kim IJ, Ku JL, Kang HC, Park JH, Yoon KA, Shin Y, Park HW, Jang SG, Lim SK, Han SY, Shin YK, Lee MR, Jeong SY, Shin HR, Lee JS, Kim WH, Park JG. 26 Mutational analysis of OGG1, MYH, MTH1 in FAP, HNPCC and sporadic colorectal cancer patients: R154H OGG1 polymorphism is associated with sporadic colorectal cancer patients. Hum Genet. 2004 Nov;115(6):498-503. 20. Soravia C, Berk T, Madlensky L, Mitri A, Cheng H, Gallinger S, Cohen Z, Bapat B. Genotype-phenotype correlations in attenuated adenomatous polyposis coli. Am J Hum Genet. 1998 Jun;62(6):1290-301. 21. Nielsen M, Hes FJ, Nagengast FM, Weiss MM, Mathus-Vliegen EM, Morreau H, Breuning MH, Wijnen JT, Tops CM, Vasen HF. Germline mutations in APC and MUTYH are responsible for the majority of families with attenuated familial adenomatous polyposis. Clin Genet. 2007 May;71(5):427-33. 22. Lynch HT, Smyrk TC. Classification of familial adenomatous polyposis: a diagnostic nightmare. Am J Hum Genet. 1998 Jun;62(6):1288-9. 23. Moisio AL, Järvinen H, Peltomäki P. Genetic and clinical characterisation of familial adenomatous polyposis: a population based study. Gut. 2002 Jun;50(6):845-50. 24. Young J, Simms LA, Tarish J, Buttenshaw R, Knight N, Anderson GJ, Bell A, Leggett B. A family with attenuated familial adenomatous polyposis due to a mutation in the alternatively spliced region of APC exon 9. Hum Mutat. 1998;11(6):450-5. 25. Rozen P, Samuel Z, Shomrat R, Legum C. Notable intrafamilial phenotypic variability in a kindred with familial adenomatous polyposis and an APC mutation in exon 9. Gut. 1999 Dec;45(6):829-33. 26. Heinimann K, Müllhaupt B, Weber W, Attenhofer M, Scott RJ, Fried M, Martinoli S, Müller H, Dobbie Z. Phenotypic differences in familial adenomatous polyposis based on APC gene mutation status. Gut. 1998 Nov;43(5):675-9. 27. van der Luijt RB, Vasen HF, Tops CM, Breukel C, Fodde R, Meera Khan P. APC mutation in the alternatively spliced region of exon 9 associated with late onset familial adenomatous polyposis. Hum Genet. 1995 Dec;96(6):705-10. 28. Kuwada SK, Burt RW, Kerber R et al. The unique phenotype of the attenuated adenomatous polyposis coli syndrome. Poster/LCPG-meeting 2001, Venice, Italy. 29. Bouguen G, Manfredi S, Blayau M, Dugast C, Buecher B, Bonneau D, 27 Siproudhis L, David V, Bretagne JF. Colorectal adenomatous polyposis Associated with MYH mutations: genotype and phenotype characteristics. Dis Colon Rectum. 2007 Oct;50(10):1612-7. 30. Wang L, Baudhuin LM, Boardman LA, Steenblock KJ, Petersen GM, Halling KC, French AJ, Johnson RA, Burgart LJ, Rabe K, Lindor NM, Thibodeau SN. MYH mutations in patients with attenuated and classic polyposis and with youngonset colorectal cancer without polyps. Gastroenterology. 2004 Jul;127(1):9-16. 31. Matsubara N, Isozaki H, Tanaka N. The farthest 3' distal end APC mutation identified in attenuated adenomatous polyposis coli with extracolonic manifestations. Dis Colon Rectum. 2000 May;43(5):720-1. 32. O'Shea AM, Cleary SP, Croitoru MA, Kim H, Berk T, Monga N, Riddell RH, Pollett A, Gallinger S. Pathological features of colorectal carcinomas in MYHassociated polyposis. Histopathology. 2008 Aug;53(2):184-94. 33. Boparai KS, Dekker E, Van Eeden S, Polak MM, Bartelsman JF, Mathus-Vliegen EM, Keller JJ, van Noesel CJ. Hyperplastic polyps and sessile serrated adenomas as a phenotypic expression of MYH-associated polyposis. Gastroenterology. 2008 Dec;135(6):2014-8. 34. Su LK, Barnes CJ, Yao W, Qi Y, Lynch PM, Steinbach G. Inactivation of germline mutant APC alleles by attenuated somatic mutations: a molecular genetic mechanism for attenuated familial adenomatous polyposis. Am J Hum Genet. 2000 Sep;67(3):582-90. 35. Brensinger JD, Laken SJ, Luce MC, Powell SM, Vance GH, Ahnen DJ, Petersen GM, Hamilton SR, Giardiello FM. Variable phenotype of familial adenomatous polyposis in pedigrees with 3' mutation in the APC gene. Gut. 1998 Oct;43(4):548-52. 36. Lynch HT, Smyrk TC, Watson P, Lanspa SJ, Lynch PM, Jenkins JX, Rouse J, Cavalieri J, Howard L, Lynch J. Hereditary flat adenoma syndrome: a variant of familial adenomatous polyposis? Dis Colon Rectum. 1992 May;35(5):411-21. 37. Couture J, Mitri A, Lagace R, Smits R, Berk T, Bouchard HL, Fodde R, Alman B, Bapat B. A germline mutation at the extreme 3' end of the APC gene results in a severe desmoid phenotype and is associated with overexpression of beta-catenin 28 in the desmoid tumor. Clin Genet. 2000 Mar;57(3):205-12. 38. Dobbie Z, Heinimann K, Bishop DT, Müller H, Scott RJ. Identification of a modifier gene locus on chromosome 1p35-36 in familial adenomatous polyposis. Hum Genet. 1997 May;99(5):653-7. 39. Järvinen H, Nyberg M, Peltokallio P. Upper gastrointestinal tract polyps in familial adenomatosis coli. Gut. 1983 Apr;24(4):333-9. 40. Iida M, Yao T, Ohsato K, Itoh H, Watanabe H. Diagnostic value of intraoperative fiberscopy for small-intestinal polyps in familial adenomatosis coli. Endoscopy. 1980 Jul;12(4):161-5. 41. DiSario J, Kuwada S, Samowitz W et al. Upper gastrointestinal lesions in the attenuated adenomatous polyposis coli syndrome (AAPC). Oral presentation/LCPG-meeting 2001, Venice, Italy. 42. Lynch HT, Smyrk TC, Lanspa SJ, Jenkins JX, Lynch PM, Cavalieri J, Lynch JF. Upper gastrointestinal manifestations in families with hereditary flat adenoma syndrome. Cancer. 1993 May 1;71(9):2709-14. 43. Hofgärtner WT, Thorp M, Ramus MW, Delorefice G, Chey WY, Ryan CK, Takahashi GW, Lobitz JR. Gastric adenocarcinoma associated with fundic gland polyps in a patient with attenuated familial adenomatous polyposis. Am J Gastroenterol. 1999 Aug;94(8):2275-81. 44. Lynch HT, Smyrk T, McGinn T, Lanspa S, Cavalieri J, Lynch J, SlominskiCastor S, Cayouette MC, Priluck I, Luce MC. Attenuated familial adenomatous polyposis (AFAP). A phenotypically and genotypically distinctive variant of FAP. Cancer. 1995 Dec 15;76(12):2427-33. 45. Friedl W, Meuschel S, Caspari R, Lamberti C, Krieger S, Sengteller M, Propping P. Attenuated familial adenomatous polyposis due to a mutation in the 3' part of the APC gene. A clue for understanding the function of the APC protein. Hum Genet. 1996 May;97(5):579-84. 46. Yanaru-Fujisawa R, Matsumoto T, Ushijima Y, Esaki M, Hirahashi M, Gushima M, Yao T, Nakabeppu Y, Iida M. Genomic and functional analyses of MUTYH in Japanese patients with adenomatous polyposis. Clin Genet. 2008 Jun;73(6):545-53. 29 47. Ponti G, Ponz de Leon M, Maffei S, Pedroni M, Losi L, Di Gregorio C, Gismondi V, Scarselli A, Benatti P, Roncari B, Seidenari S, Pellacani G, Varotti C, Prete E, Varesco L, Roncucci L. Attenuated familial adenomatous polyposis and MuirTorre syndrome linked to compound biallelic constitutional MYH gene mutations. Clin Genet. 2005 Nov;68(5):442-7. 48. Ali M, Kim H, Cleary S, Cupples C, Gallinger S, Bristow R. Characterization of mutant MUTYH proteins associated with familial colorectal cancer. Gastroenterology. 2008 Aug;135(2):499-507. 49. Peters U, Chatterjee N, Hayes RB, Schoen RE, Wang Y, Chanock SJ, Foster CB. Variation in the selenoenzyme genes and risk of advanced distal colorectal adenoma. Cancer Epidemiol Biomarkers Prev. 2008 May;17(5):1144-54. 50. Croce CM. Oncogenes and cancer. N Engl J Med. 2008 Jan 31;358(5):502-11. 51. Goss KH, Groden J. Biology of the adenomatous polyposis coli tumor suppressor. J Clin Oncol. 2000 May;18(9):1967-79. 52. Näthke I. APC at a glance. J Cell Sci. 2004 Oct 1;117(Pt 21):4873-5. 53. Spirio L, Green J, Robertson J, Robertson M, Otterud B, Sheldon J, Howse E, Green R, Groden J, White R, Leppert M. The identical 5' splice-site acceptor mutation in five attenuated APC families from Newfoundland demonstrates a founder effect. Hum Genet. 1999 Nov;105(5):388-98. 54. Bunyan DJ, Shea-Simonds J, Reck AC, Finnis D, Eccles DM. Genotypephenotype correlations of new causative APC gene mutations in patients with familial adenomatous polyposis. J Med Genet. 1995 Sep;32(9):728-31. 55. Nasioulas S, Jones IT, St John DJ, Scott RJ, Forrest SM, McKinlay Gardner RJ. Profuse familial adenomatous polyposis with an adenomatous polyposis coli exon 3 mutation. Fam Cancer. 2001;1(1):3-7. 56. Dietrich WF, Lander ES, Smith JS, Moser AR, Gould KA, Luongo C, Borenstein N, Dove W. Genetic identification of Mom-1, a major modifier locus affecting Min-induced intestinal neoplasia in the mouse. Cell. 1993 Nov 19;75(4):631-9. 57. Slupska MM, Baikalov C, Luther WM, Chiang JH, Wei YF, Miller JH. Cloning and sequencing a human homolog (hMYH) of the Escherichia coli mutY gene whose function is required for the repair of oxidative DNA damage. J Bacteriol. 30 1996 Jul;178(13):3885-92. 58. Yamaguchi S, Shinmura K, Saitoh T, Takenoshita S, Kuwano H, Yokota J. A single nucleotide polymorphism at the splice donor site of the human MYH base excision repair genes results in reduced translation efficiency of its transcripts. Genes Cells. 2002 May;7(5):461-74. 59. Croitoru ME, Cleary SP, Berk T, Di Nicola N, Kopolovic I, Bapat B, Gallinger S. Germline MYH mutations in a clinic-based series of Canadian multiple colorectal adenoma patients. J Surg Oncol. 2007 May 1;95(6):499-506. 60. Sieber OM, Lipton L, Crabtree M, Heinimann K, Fidalgo P, Phillips RK, Bisgaard ML, Orntoft TF, Aaltonen LA, Hodgson SV, Thomas HJ, Tomlinson IP. Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N Engl J Med. 2003 Feb 27;348(9):791-9. 61. Cleary SP, Cotterchio M, Jenkins MA, Kim H, Bristow R, Green R, Haile R, Hopper JL, LeMarchand L, Lindor N, Parfrey P, Potter J, Younghusband B, Gallinger S. Germline MutY human homologue mutations and colorectal cancer: a multisite case-control study. Gastroenterology. 2009 Apr;136(4):1251-60. 62. Sampson JR, Dolwani S, Jones S, Eccles D, Ellis A, Evans DG, Frayling I, Jordan S, Maher ER, Mak T, Maynard J, Pigatto F, Shaw J, Cheadle JP. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet. 2003 Jul 5;362(9377):39-41. 63. Lipton L, Tomlinson I. The multiple colorectal adenoma phenotype and MYH, a base excision repair gene. Clin Gastroenterol Hepatol. 2004 Aug;2(8):633-8. 64. Di Gregorio C, Frattini M, Maffei S, Ponti G, Losi L, Pedroni M, Venesio T, Bertario L, Varesco L, Risio M, Ponz de Leon M. Immunohistochemical expression of MYH protein can be used to identify patients with MYH-associated polyposis. Gastroenterology. 2006 Aug;131(2):439-44. 65. Nielsen M, Joerink-van de Beld MC, Jones N, Vogt S, Tops CM, Vasen HF, Sampson JR, Aretz S, Hes FJ. Analysis of MUTYH genotypes and colorectal phenotypes in patients With MUTYH-associated polyposis. Gastroenterology. 2009 Feb;136(2):471-6. 66. Sampson JR, Dolwani S, Jones S, Eccles D, Ellis A, Evans DG, Frayling I, 31 Jordan S, Maher ER, Mak T, Maynard J, Pigatto F, Shaw J, Cheadle JP. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet. 2003 Jul 5;362(9377):39-41. 67. Venesio T, Molatore S, Cattaneo F, Arrigoni A, Risio M, Ranzani GN. High frequency of MYH gene mutations in a subset of patients with familial adenomatous polyposis. Gastroenterology. 2004 Jun;126(7):1681-5. 68. Tricarico R, Bet P, Ciambotti B, Di Gregorio C, Gatteschi B, Gismondi V, Toschi B, Tonelli F, Varesco L, Genuardi M. Endometrial cancer and somatic G>T KRAS transversion in patients with constitutional MUTYH biallelic mutations. Cancer Lett. 2009 Feb 18;274(2):266-70. 69. Nance FC. Management strategies for familial adenomatous polyposis. Ann Surg. 1993 Feb;217(2):99-100. 70. Lynch HT, Lynch JF, Lynch PM, Attard T. Hereditary colorectal cancer syndromes: molecular genetics, genetic counseling, diagnosis and management. Fam Cancer. 2008;7(1):27-39. 71. Ponti G, Ponz de Leon M. Muir-Torre syndrome. Lancet Oncol. 2005 Dec;6(12):980-7. 72. King JE, Dozois RR, Lindor NM, Ahlquist DA. Care of patients and their families with familial adenomatous polyposis. Mayo Clin Proc. 2000 Jan;75(1):57-67. 73. Moisio AL, Järvinen H, Peltomäki P. Genetic and clinical characterisation of familial adenomatous polyposis: a population based study. Gut. 2002 Jun;50(6):845-50. 74. Fornasarig M, Minisini AM, Viel A, Quaia M, Canzonieri V, Veronesi A. Twelve years of endoscopic surveillance in a family carrying biallelic Y165C MYH defect: report of a case. Dis Colon Rectum. 2006 Feb;49(2):272-5. 75. Hampel H, Peltomaki P. Hereditary colorectal cancer: risk assessment and management. Clin Genet. 2000 Aug;58(2):89-97. 76. Bülow C, Vasen H, Järvinen H, Björk J, Bisgaard ML, Bülow S. Ileorectal anastomosis is appropriate for a subset of patients with familial adenomatous polyposis. Gastroenterology. 2000 Dec;119(6):1454-60. 32 77. Church J, Burke C, McGannon E, Pastean O, Clark B. Risk of rectal cancer in patients after colectomy and ileorectal anastomosis for familial adenomatous polyposis: a function of available surgical options. Dis Colon Rectum. 2003 Sep;46(9):1175-81. 78. Lynch HT, Watson P. AFAP: variety is the spice of life. Gut. 1998 Oct;43(4):451-2. 79. Vasen HF, van der Luijt RB, Slors JF, Buskens E, de Ruiter P, Baeten CG, Schouten WR, Oostvogel HJ, Kuijpers JH, Tops CM, Meera Khan P. Molecular genetic tests as a guide to surgical management of familial adenomatous polyposis. Lancet. 1996 Aug 17;348(9025):433-5. 80. Rozen P, Macrae F. Familial adenomatous polyposis: The practical applications of clinical and molecular screening. Fam Cancer. 2006;5(3):227-35. 81. Waddell WR, Ganser GF, Cerise EJ, Loughry RW. Sulindac for polyposis of the colon. Am J Surg. 1989 Jan;157(1):175-9. 82. Phillips RK, Wallace MH, Lynch PM, Hawk E, Gordon GB, Saunders BP, Wakabayashi N, Shen Y, Zimmerman S, Godio L, Rodrigues-Bigas M, Su LK, Sherman J, Kelloff G, Levin B, Steinbach G; FAP Study Group. A randomised, double blind, placebo controlled study of celecoxib, a selective cyclooxygenase 2 inhibitor, on duodenal polyposis in familial adenomatous polyposis. Gut. 2002 Jun;50(6):857-60. 83. Giardiello FM, Hamilton SR, Krush AJ, Piantadosi S, Hylind LM, Celano P, Booker SV, Robinson CR, Offerhaus GJ. Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N Engl J Med. 1993 May 6;328(18):1313-6. 84. Lynch HT, Thorson AG, Smyrk T. Rectal cancer after prolonged sulindac chemoprevention. A case report. Cancer. 1995 Feb 15;75(4):936-8. 85. Bresalier RS, Sandler RS, Quan H, Bolognese JA, Oxenius B, Horgan K, Lines C, Riddell R, Morton D, Lanas A, Konstam MA, Baron JA; Adenomatous Polyp Prevention on Vioxx (APPROVe) Trial Investigators. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005 Mar 17;352(11):1092-102. 33 86. Wallace MH, Phillips RK. Upper gastrointestinal disease in patients with familial adenomatous polyposis. Br J Surg. 1998 Jun;85(6):742-50. 87. Gallagher MC, Phillips RK, Bulow S. Surveillance and management of upper gastrointestinal disease in Familial Adenomatous Polyposis. Fam Cancer. 2006;5(3):263-73. 88. Wallace MH, Lynch PM. The current status of chemoprevention in FAP. Fam Cancer. 2006;5(3):289-94; discussion 295-6. 89. Laken SJ, Papadopoulos N, Petersen GM, Gruber SB, Hamilton SR, Giardiello FM, Brensinger JD, Vogelstein B, Kinzler KW. Analysis of masked mutations in familial adenomatous polyposis. Proc Natl Acad Sci U S A. 1999 Mar 2;96(5):2322-6. 90. Ponz de Leon M. Hereditary Gastrointestinal Polyposis Syndrome. In: “Familial and Hereditary Tumors”. Springer-Verlag, Heidelberg, 1994;238-64. 91. Lynch HT, Tinley ST, Lynch J, Vanderhoof J, Lemon SJ. Familial Adenomatous Polyposis: discovery of a family and its management in a Cancer Genetics Clinic. Cancer 1997;80:614-20. 92. Utsunomiya J and Iwama T. Adenomatosis Coli in Japan. Colorectal Cancer: Prevention, Epidemiology, and Screening. 1980:83-95. 93. Kinney AY, Hicken B, Simonsen SE, Venne V, Lowstuter K, Balzotti J, Burt RW. Colorectal cancer surveillance behaviors among members of typical and attenuated FAP families. Am J Gastroenterol. 2007 Jan;102(1):153-62. 94. Sampson JR, Dolwani S, Jones S, Eccles D, Ellis A, Evans DG, Frayling I, Jordan S, Maher ER, Mak T, Maynard J, Pigatto F, Shaw J, Cheadle JP. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet. 2003 Jul 5;362(9377):39-41. 95. Crabtree MD, Fletcher C, Churchman M, Hodgson SV, Neale K, Phillips RK, Tomlinson IP. Analysis of candidate modifier loci for the severity of colonic familial adenomatous polyposis, with evidence for the importance of the N-acetyl transferases. Gut. 2004 Feb;53(2):271-6. 96. Crabtree MD, Tomlinson IP, Hodgson SV, Neale K, Phillips RK, Houlston RS. Explaining variation in familial adenomatous polyposis: relationship between 34 genotype and phenotype and evidence for modifier genes. Gut. 2002 Sep;51(3):420-3. 97. Wallace MH, Lynch PM. The current status of chemoprevention in FAP. Fam Cancer. 2006;5(3):289-94; discussion 295-6. 98. Wakai K, Date C, Fukui M, Tamakoshi K, Watanabe Y, Hayakawa N, Kojima M, Kawado M, Suzuki K, Hashimoto S, Tokudome S, Ozasa K, Suzuki S, Toyoshima H, Ito Y, Tamakoshi A; JACC Study Group. Dietary fiber and risk of colorectal cancer in the Japan collaborative cohort study. Cancer Epidemiol Biomarkers Prev. 2007 Apr;16(4):668-75. 99. Koushik A, Hunter DJ, Spiegelman D, Beeson WL, van den Brandt PA, Buring JE, Calle EE, Cho E, Fraser GE, Freudenheim JL, Fuchs CS, Giovannucci EL, Goldbohm RA, Harnack L, Jacobs DR Jr, Kato I, Krogh V, Larsson SC, Leitzmann MF, Marshall JR, McCullough ML, Miller AB, Pietinen P, Rohan TE, Schatzkin A, Sieri S, Virtanen MJ, Wolk A, Zeleniuch-Jacquotte A, Zhang SM, Smith-Warner SA. Fruits, vegetables, and colon cancer risk in a pooled analysis of 14 cohort studies. J Natl Cancer Inst. 2007 Oct 3;99(19):1471-83. 100. Pedersen A, Johansen C, Grønbaek M. Relations between amount and type of alcohol and colon and rectal cancer in a Danish population based cohort study. Gut. 2003 Jun;52(6):861-7. 101. Terry MB, Neugut AI, Bostick RM, Sandler RS, Haile RW, Jacobson JS, Fenoglio-Preiser CM, Potter JD. Risk factors for advanced colorectal adenomas: a pooled analysis. Cancer Epidemiol Biomarkers Prev. 2002 Jul;11(7):622-9. 102. Michels KB, Giovannucci E, Chan AT, Singhania R, Fuchs CS, Willett WC. Fruit and vegetable consumption and colorectal adenomas in the Nurses' Health Study. Cancer Res. 2006 Apr 1;66(7):3942-53. 103. Tsubono Y, Otani T, Kobayashi M, Yamamoto S, Sobue T, Tsugane S; JPHC Study Group. No association between fruit or vegetable consumption and the risk of colorectal cancer in Japan. Br J Cancer. 2005 May 9;92(9):1782-4. 104. Umetani N, Watanabe T, Sasaki S, Nagawa H. Rectosigmoidal adenomatous polyposis: a novel entity of polyposis? Report of a case. Dis Colon Rectum. 2000 Oct;43(10):1439-43. 35 105. De Pietri S, Sassatelli R, Roncucci L, Bertoni G, Landi P, Sabadini G, Tansini P, Cavallini G, Cantoni E, Mareni C, et al. Clinical and biologic features of adenomatosis coli in Northern Italy. Scand J Gastroenterol. 1995 Aug;30(8):7719. 106. Truta B, Allen BA, Conrad PG, Weinberg V, Miller GA, Pomponio R, Lipton LR, Guerra G, Tomlinson IP, Sleisenger MH, Kim YS, Terdiman JP. A comparison of the phenotype and genotype in adenomatous polyposis patients with and without a family history. Fam Cancer. 2005;4(2):127-33. 36 DESCRIPTION OF THE FAMILIES WITH ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS OBSERVED IN MODENA BETWEEN 1984 AND 2009 In more than 25 years of continuous activity, the Colorectal Cancer Study Group of the University of Modena and Reggio Emilia examined several hundred of pedigrees, either of patients diagnosed through the local Colorectal Cancer Registry or individuals addressed to the Institution from other Provinces and Regions. In 28 of these pedigrees (often single individuals) features of AFAP were observed in the proband and, in some cases, in other members of the family. In particular, a total of 10 to 99 synchronous adenomas in the large bowel. These cases have been classified as “Attenuated Familial Adenomatous Polyposis (AFAP)”. In 5 of these families constitutional mutations of the APC gene (n. 3) or MutYH gene (n. 2) were detected. For each family a genealogical tree has been traced, as it appeared at the last visit (usually between 2007 and 2009). Below the pedigree there is a table which summarizes, for that specific proband and family, the most relevant clinical data. The second page shows a schematic family history, that reports the most significant diseases of the proband and other affected family members. Emphasis has been given to benign and malignant neoplasms of the large bowel. When information was scanty, lacking or incomplete, this has been indicated in the text. Finally, in a box at the base of the second page we described the results of genetic testing carried out on constitutional DNA (i.e., search for APC and MutYH mutations). 37 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) N.1 2 1 I K colon 78 a 86 1 II AFAP + K colon 65 1 2 3 III a 55 a 43 a 49 IV a 31 a 18 Patient Diagnosis Age Extracolonic Manifestations Other relevant Diseases Surgery II-1 AFAP + Cancer of the Cecum 65 _ 1st operation Subtotal colectomy and ileorectal anastomosis (IRA) (1989) 2nd operation Proctectomy and Ileostomy (Recurrence, 1994) Mutations (APC/MutYH) Not tested 38 History of the proband and the family. The proband (II-1) underwent colonoscopy in 1989, at the age of 65 years, because of abdominal pain and bloating. The investigation showed diffuse polyposis especially in the transverse and ascending colon, with polyps ranging from 2-3 mm to 4 cm in diameter. A malignant lesion occupied the cecum. After a few days, the patient was operated on of subtotal colectomy and IRA. At anatomical examination a total of 78 adenomas with different degrees of dysplasia could be observed in various segments of the large bowel, and an adenocarcinoma (Stage III TNM, Dukes’ C) of the cecum. After the operation, the patient did not undergo chemotherapy; he was reluctant to be followed by rectoscopy at regular interval of time. In 1994 he noticed rectal bleeding, and a neoplasm of the rectal stump was diagnosed. The patient was operated on of proctectomy and permanent ileostomy. After the operation the patient did not complain other troubles related to the disease. He died in 2003, at age 79, owing to complications of pneumonia. In the family, the father of the proband was operated on at the age of 78 years for cancer of the large bowel, and died after a few months. No other cases of neoplasms were referred. The 3 daughters of the proband refused (and continue to refuse) any investigation. Molecular characteration No genetic test has been carried out. 39 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) N. 2 1 2 I a 72 a 69 1 II AFAP 35 1 III a 11 Patient Diagnosis Age Extracolonic Manifestations Other relevant Diseases Surgery II-1 AFAP 35 _ _ Mutations (APC/MutYH) Negative 40 History of the proband and the family. After several years of abdominal discomfort attributed to “irritable bowel”, the proband underwent colonoscopy in 2002, at the age of 35 years. The endoscopic description was rather vague (“numerous polyps” in various segments of the large bowel; the number was less than 100), however, 30 polyps were removed and analyzed. The histological examination showed tubular and tubulovillous adenomas with low-grade dysplasia. The search for extracolonic manifestations was negative. From 2002 to 2007 the patient was treated with Celecoxilb (200 mg x 2). Various endoscopic controls documented the reoccurrence of 1 to a few small polyps, which were removed and analyzed (usually tubular adenomas with low-grade dysplasia). At the last control (April 2008), 1 small adenoma and two hyperplastic polyps were observed and removed. No other member of the family showed features of polyposis. Molecular characteration No constitutional alterations of the APC and MutYH genes were detected. 41 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) N. 3 2 1 I a 80 1 a 84 2 3 4 II a 61 AFAP + K colon 49 a 58 1 2 3 4 5 a 35 a 24 a 35 a 33 a 24 a 49 6 III a 20 Patient Diagnosis Age Extracolonic Manifestations Other relevant Diseases Surgery Mutations (APC/MutYH) II-3 AFAP + Cancer of the Ascending colon 49 Thyroid Cyst Subtotal colectomy and IRA Negative 42 History of the proband and the family. At the age of 49 years, the proband, II-3, complained severe and persistent abdominal pain with no other relevant symptoms. A subsequent colonoscopy showed the presence of numerous polyps in the large bowel and an infiltrating lesion in the ascending colon. In 2003 (age 50) the patient was operated on of subtotal colectomy and ileorectal anastomosis. At anatomical examination a total of 22 polyps (adenomas, with various degrees of dysplasia) were detected. In the ascending colon a Dukes C adenocarcinoma (T3N2M0) could be observed. The patient received 5 FU-based chemotherapy for 6 months. After surgery, the endoscopic follow-up has been so far unremarkable. No other member of the family showed features of polyposis. In particular, two out of 3 siblings (II1 and II-2) underwent colonoscopy, which resulted negative. The two offsprings of the proband (III5 and III-6) have not been tested. Both the parents of the proband (I-1 and I-2) died over the age of 80 years for causes unrelated to neoplasms. Molecular characteration No mutations were detected in the APC and MutYH genes. 43 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) N. 4 1 2 I a 79 1 a 87 2 3 II AFAP 47 1 AFAP +K colon 43 3 2 4 5 III a 30 a 31 a 24 a 21 Patient Diagnosis Age Extracolonic Manifestations Other relevant Diseases Surgery Mutations (APC/MutYH) II-1 AFAP 47 _ _ MutYH + II-3 AFAP + K Colon 43 _ Subtotal colectomy and IRA MutYH + a 18 t 44 History of the proband and the family. The proband (II-1) underwent colonoscopy in 1998, at the age of 47 years, when she knew that the brother had been operated on for colorectal cancer. The endoscopy showed the presence of nearly 50 small polyps scattered in the various segments of the large bowel. The lesions were subsequently all removed, and histological examination revealed tubular adenomas with low-medium degree dysplasia. Since then the patient underwent regular endoscopic controls with removal of the newly developed lesions. The last two colonoscopies (2007 and 2008) were negative. The proband’s brother (II-3) complained symptoms of abdominal discomfort in early 1998, at the age of 43 years. An endoscopic evaluation raised the suspicion of malignant lesions in the sigmoid colon, and the patient was operated on of subtotal colectomy and ileorectal anastomosis. At pathology, 2 malignant lesions were present (sigmoid and splenic flexure; T3N0M0, T1N0M0), together with 11 adenomatous polyps mainly located in the left colon. Subsequent endoscopic controls showed the recurrence of a few micropolyps in the rectal stump. No other member of the family showed features of polyposis. Lower endoscopies were normal in the two offsprings of the proband. The most interesting aspect of the family was the molecular characterization, the proband being heterozygous and the proband’s brother homozygous for the same MutYH mutation. Molecular characteration Mutation detected Gene: MutYH Exon: 14 Type of alteration: 1395 del GGA/wt (proband) 1395 del GGA/1395 del GGA (II-3) Functional change: Removal of a glutamic acid within a highly conserved region of the protein. 45 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) N. 5 2 1 I a 85 K uterus 81 1 2 II AFAP 50 AFAP 50 1 2 III a 36 a 35 Patient Diagnosis Age Extracolonic Manifestations Other relevant Diseases Surgery Mutations II-1 AFAP 50 Sebaceous lesion of the skin Right Hemicolectomy MutYH + II-2 AFAP 50 Gastric Polyps Thyroid Nodule Colectomy and IRA MutYH + 46 History of the proband and the family. In 2000, the proband (II-2) complained rectal bleeding and abdominal pain, at the age of 50 years. Colonoscopy disclosed the presence of numerous polyps in the various large bowel tracts. In the same year, the patient underwent colectomy and ileorectal anastomosis. At anatomical examination a total of 24 polyps (tubular and tubulovillous adenomas with low-medium grade dysplasia) were detected. At subsequent follow-up, small polyps (hyperplastic) of the rectal stump were removed. The first upper endoscopy showed the presence of glandular polyps of the stomach (2004). During follow-up a large adenoma (3 cm) of the gastric fundus was detected and removed (2008) through endoscopy (tubulovillous adenoma with medium grade dysplasia). At both examinations the duodenum was normal. A small thyroid nodule (6 mm) was observed in 2003 (not biopsed). The brother of the proband (II-1) underwent colonoscopy in 1996 owing to aspecific abdominal symptoms. Some 25-30 polyps were detected, predominantly localized in the proximal colon, and the patient was operated on of right hemicolectomy (1996). Small polyps (hyperplastic and adenomatous) were detected during surveillance of the remaining colon (2000, 2003, 2005). Small sebaceous lesions were removed from the face (2005). The proband’s mother died of cancer of the uterus (not otherwise specified). Molecular characteration Mutation detected Gene: MutYH Exon: 7 Type of alteration: Y165C/G382D (compound heterozygosis) Functional change: Reduced activity of the MutYH protein (Y165C) ; Amino Acid change (G382D). 47 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) N. 6 1 2 I a 85 1 2 a 96 4 3 5 II a 61 AFAP + K cecum 60 a 71 2 1 4 a 67 a 56 5 6 3 III a 37 0 a 41 a 44 a 42 a 36 a 34 Patient Diagnosis Age Extracolonic Manifestations Other relevant Diseases Surgery Mutations (APC/MutYH) II-3 AFAP + K Cecum 60 None Subtotal Colectomy and IRA Negative (MutYH) In progress (APC) 48 History of the proband and the family. The proband (II-3) underwent colonoscopy in 2000, at the age of 60 years, because of abdominal discomfort and anaemia; the endoscopist described the presence of about 50 polyps, both sessile and pedunculated, ranging between 0.5 and 1.5 cm in diameter, distributed along the various tracts of the large bowel. In the splenic flexure a vegetating mass of 6.0 cm of diameter suspected of malignancy was noted. After a few months, the patient was operated on of subtotal colectomy and IRA (December 2000). At anatomical examination, 30 lesions were analyzed; the majority of them were adenomatous polyps with various degrees of dysplasia. In the splenic flexure, a TNM I (T2N0M0, Dukes A) adenocarcinoma was diagnosed. At least two successive endoscopic controls resulted negative. Similarly, the search for extracolonic changes (osteomas, retinal spots, gastric and duodenal polyps) was negative. From 2005 we lost the patient from follow-up. No other member of the family showed features of polyposis, although sporadic adenomas were removed during colonoscopy in patients II-2 and II-5. Molecular characteration Search for mutation in the MutYH gene: negative. Study of APC gene in process. 49 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) N. 7 1 2 I a 91 K pancreas 83 1 2 II K uterus 45 AFAP + K duodenum 59 1 2 a 46 a 39 III a 30 Patient Diagnosis Age Extracolonic Manifestations Other relevant Diseases Surgery II-2 AFAP 59 Duodenal Cancer age 59 1st operation Subtotal Colectomy and IRA 2nd operation Duodenocefalopancreasectomy Mutations (APC/MutYH) Negative 50 History of the proband and the family. In 2005, at the age of 59 years, the proband of this family (II-2) complained persistent abdominal pain, which was followed by jaundice. The patient was hospitalised and underwent a series of investigations. An upper abdomen ultrasound study showed the presence of multiple stones in the gallbladder and bile duct; moreover, gastroscopy revealed a large (5x7 cm) polyp of the duodenum, extended to the entire circumference around the papilla of Vater. Another polyp (2 cm) protruded from the papilla. Colonoscopy showed the presence of nearly 20 polyps in the various segments of the large bowel, ranging between 1 and 3 cm in diameter. Bile duct stones were treated successfully by endoscopy; the patient was subsequently operated on (2005) of subtotal colectomy and ileorectal anastomosis. At anatomical description “numerous” (between 30 and 50 according to the wife) polyps were observed of various dimensions (tubular adenomas with villous component and moderate to severe dysplasia but without evidence of infiltrating malignancy). After a few months (November 2005), the patient was operated on of duodeno-cefalo-pancreasectomy (the polyps could not be removed endoscopically). Histology could not be obtained, but malignant changers were likely. In fact, after 3 years the patient developed hepatic metastases and died within a few months despite chemotherapy. No other extracolonic manifestations could be detected. In the family, the sister of the proband was operated on for a carcinoma for the uterus (not otherwise specified) at the age of 45 years; she is at present in good health at age 68. The father of the proband died of pancreatic cancer at age 83. As far as we know, no other member of the family showed colorectal polyposis. Molecular characteration No MutYH or APC mutations were detected. 51 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) N. 8 1 2 I 1 II 2 3 4-6 7 3 1 AFAP+ K colon 72 AFAP? K(?) 1 2-11 12-14 3 10 III Patient Diagnosis Age Extracolonic Manifestations Other relevant Diseases Surgery II-1 AFAP + K Colon 72 _ Subtotal Colectomy and IRA Mutations (APC/MutYH) Not Tested 52 History of the proband and the family. The information obtained from this family is fragmentary and insufficient for several reasons, including poor collaboration. The proband (II-1) was affected from the birth by a severe mental disease (not otherwise specified) and spent her life in a hospice. In 1987, at the age of 72, she was hospitalized because of severe abdominal pain and rectal bleeding. At lower endoscopy, numerous small sessile polyps were detected in the sigmoid, descending, transverse and ascending colon. After one month, the patient was operated on of subtotal colectomy and ileorectal anastomosis. At anatomical examination, nearly 50 sessile and peduncolated polyps were observed, ranging from few mm to 1.5 cm. At histology, most of the lesions were tubular or villous adenomas with various degrees of dysplasia; the largest polyps (sigmoid) showed features of infiltrative neoplasm (Dukes A-TNM1 adenocarcinoma). The patient underwent endoscopic controls for the successive four years, with removal of several newly developed adenomas. Rather unexpectedly, the patient died of local recurrence in 1998 at the age of 83 years. It was referred by the family doctor that a brother of the proband (II-2) was affected by polyposis, but the finding has never been documented, and we have not been able to contact the patient or his family. Another sibling presumably died of cancer but, again, confirmation was not obtained. Since patients II-1 and II-2 had no offsprings, the disease presumably faded with them. Molecular characteration No molecular analyzes could be carried out in this family. 53 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) N. 9 1 2 I a 69 a 60 1 2 II AFAP 18 a 18 III Patient Diagnosis Age Extracolonic Manifestations Other relevant Diseases Surgery II-1 AFAP 18 - Osteomas - Gastric polyps - Duodenal polyps _ Mutations (APC/MutYH) APC + 54 History of the proband and the family. The onset of the disease in this family was rather anomalous. In 2006, at the age of 18 years, the proband (II-1) complained a certain difficulty in mastication, which was attributed to mandibular osteomas (documented through a radiogram). In the suspicion of polyposis/Gardener syndrome, the patient underwent lower endoscopy, that showed the presence of around 30 polyps of 3 to 5 mm in diameter in the ascending colon, sigmoid and rectum. Histological examination of 10 removed polyps revealed features of tubular adenoma with low-grade dysplasia. The patient started treatment with Celecoxib (200 mg/day); subsequent endoscopies (2007, 2008) confirmed the presence of polyps, which appeared unchanged in size, number and histological features. A gastroduodenoscopy (2006) showed the stomach carpeted by small lesions (glandular polyps, 2-4 mm in diameter) and a normal duodenum. At a subsequent gastroscopy, the stomach appeared unmodified, while numerous polyps of 2-3 mm in size appeared in the duodenum (tubular adenomas, low-grade dysplasia). Both the proband’s parents underwent colonoscopy, which was unremarkable. Molecular characteration Mutation detected Gene: APC Exon: 15 Type of alteration: 4612 del GA Functional change: formation of a stop codon (truncated protein) 55 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) N. 10 1 2 I a 82 a 81 2 1 3 II AFAP+ K rectum 40 a 39 a 59 1 2 3 III a 21 a 15 a5 Patient Diagnosis Age Extracolonic Manifestations Other relevant Diseases Surgery Mutations (APC/MutYH) II-2 AFAP + Cancer of the Rectum 40 _ Anterior Resection of the Rectum Negative (MutYH) In progress (APC) 56 History of the proband and the family. The proband (II-2) was operated on for rectal carcinoma (Dukes C) in 2001, at the age of 40 years, owing to rectal bleeding. At anatomical examination, “numerous” (4) polyps were seen around the tumor mass, ranging from 0.6 to 1.0 cm in diameter (tubular adenomas, with low medium-grade dysplasia). In the subsequent years the patient underwent several colonoscopies, which revealed the presence of one or more small adenomas. In one of these controls (2005) more than 20 small adenomas in the sigmoid and descending colon were observed and biopsed. The patient is still under endoscopic control, with the constant removal of the lesions larger than 10 mm and the careful observation of the smaller polyps. In the family, only subject II-1 (proband’s brother) underwent colonoscopy, which showed the presence of a few small polyps (removed). Apparently, this is an unusual family in which rectal carcinoma developed some years before the appearance of attenuated polyposis Molecular characteration Absence of Microsatellite Instability (BAT25, BAT26, CAT25) Search for constitutional mutations of MutYH gene: negative. 57 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) N. 11 1 2 I K colon 68 1 2 a > 70 3-5 II 3 3 K colon 63 K lung 78 AFAP 62 7 3 III 9 3 6-8 3 a 32 Patient Diagnosis Age Extracolonic Manifestations Other relevant Diseases Surgery II-9 AFAP 62 _ _ a 28 Mutations (APC/MutYH) Negative (MutYH) In progress (APC) 58 History of the proband and the family. At the age of 48 years, the proband underwent lower endoscopy because of rectal bleeding; a small (5 mm) polyp of the sigmoid was found. Subsequent endoscopies (1996, 2001) were negative but, in 2007, twenty micropolyps were observed and removed. Anatomical examination revealed tubular or tubulovillous adenomas with various degrees of dysplasia. Six more adenomatous lesions were removed in 2008, at age 63. No surgical operation was taken into consideration, and the patient continued with endoscopic follow-up. Two components of the family (II-1, proband’s sister and I-1 proband’s father) died of colorectal cancer (document in the sister, by history in the father), but apparently without features of polyposis. Another sibling of the proband (II-2) died of lung cancer at age 78. The case is of interest, mainly because there is evidence of polyp appearance at the age of 62, whereas previous endoscopies did not show features of polyposis. Molecular characteration Search of MutYH mutations: negative. APC gene analysis: in process. 59 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) N. 12 1 2 I a 75 K adrenal gland 65 2 1 3 II 1 a 50 a 54 AFAP 41 2 3 4 5 6 a 23 a 21 a 15 a 25 a 22 III a 28 Patient Diagnosis Age Extracolonic Manifestations Other relevant Diseases II-1 AFAP 41 _ Surgery _ Mutations (APC/MutYH) Negative I 60 History of the proband and the family. Because of abdominal discomfort, the proband (II-1) underwent colonoscopy in 1996, at the age 41 years. This revealed the presence of numerous polyps in the transverse, descending and sigmoid colon. A total of 42 lesions were removed and analyzed (2-5 mm in diameter, tubular adenomas with low-grade dysplasia; in only one of the adenomas an area of severe dysplasia was observed). Other small adenomas were removed during the numerous endoscopies carried out between 1997 and 2007 (the colonoscopy has never been “negative”). The search for extracolonic manifestations (retinal spots, osteomas, dental abnormalities, gastric and duodenal polyps) was negative. A thyroid adenoma (2.1 cm in diameter) was diagnosed in 1997. At the last control (February 2009) a total of 10 lesions were removed from the transverse, descending, sigmoid and rectum, 3 to 5 mm in diameter (tubular adenomas with low grade dysplasia). No other member of the family showed features of polyposis. The father of the proband, however, underwent the removal of an adrenal gland tumor at the age of 65 years, and a few years later polyps were removed from the bladder. In the older son of the proband (III-1), colonoscopy was negative. Molecular characteration No APC or MutYH mutations were detected. 61 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) N. 13 1 2 I K colon 70 1 a 86 2 3 4 II AFAP+ K colon 47 1 2 AFAP+ K colon 47 a 58 3 4 5 AFAP 39 6 7 a 18 a 16 III a 37 a 34 a 19 a 16 a 11 Patient Diagnosis Age Extracolonic Manifestations Other relevant Diseases Surgery Mutations (APC/MutYH) II- 1 AFAP + K Colon 47 _ _ Not tested II- 3 AFAP + K Colon 47 _ _ Not tested III- 4 AFAP K Colon 39 46 _ Left Hemicolectomy (age 46) Negative 62 History of the proband and the family. The proband (II-4) underwent lower endoscopy in 2000, at the age of 39 years, upon suggestion of the family doctor (after the death of her brother). In the transverse, descending and rectosigmoid colon a total of 30 polyps were observed, ranging from 3 mm to 2.0 cm in diameter, many of which were removed (tubular adenomas with moderate dysplasia). The patient went on with irregular endoscopic controls. Surgery was suggested, but the proband postponed the decision and chose to be cured with alternative/complementary remedies (Achillea, Ginkgo, Hypericus and others). At the last endoscopy, the suspicion of malignancy in one of the largest polyps was raised; the patient was operated on of left hemicolectomy as her own choice (the surgeon had suggested colectomy and ileorectal anastomosis), in 2007. The anatomical examination confirmed the presence of a TNM II adenocarcinoma of the sigmoid. The search for extracolonic manifestation was negative (retinal spots, gastric or duodenal polyps, osteomas). Information on the family was scanty; two of the 3 siblings were affected by colorectal neoplasia both at the age of 47 years, presumably over a background of polyposis, and both died shortly after diagnosis. The proband’s father (I-1) died of colorectal cancer at age 70, but no further information was available. It is tempting to speculate that a deleterious mutation transmitted vertically from the father to three of the offsprings was present; however, no constitutional alteration has so far been detected. Molecular characteration No mutations in the APC or MutYH genes were detected. 63 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) N. 14 1 2 I a 53 a 85 1 2 II a 61 AFAP 54 2 3 a 28 a 23 1 III a 30 Patient Diagnosis Age Extracolonic Manifestations Other relevant Diseases Surgery II-2 AFAP 54 _ _ Mutations (APC/MutYH) In progress 64 History of the proband and the family. The proband executed a fecal occult blood test within a screening program of the Region EmiliaRomagna. Since the search for occult blood was positive, he underwent colonoscopy; this revealed the presence of nearly 30 polyps, from 0.5 to 3.0 cm, in the rectal ampulla (the largest), in the sigmoid and transverse colon. All the lesions were removed during two successive endoscopies (2006, at the age of 54 years). At histological examination, features of tubular or tubulo-villous adenomas were seen in all removed lesions. Three micropolyps were taken off in a subsequent endoscopy, in 2007. An upper gastrointestinal endoscopy (2008) showed mild esophagitis. The last colonoscopy (2008) was unremarkable; in the same year a blood sample was taken for genetic tests. No other member of the family showed features of polyposis. Molecular characteration Analysis of APC and MutYH genes in progress. 65 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) N. 15 1 I 2 K lung 81 K prostate 81 a 81 1 II 2 AFAP + K colon 52 a 61 1 III a 26 Patient Diagnosis Age Extracolonic Manifestations Other relevant Diseases Surgery Mutations (APC/MutYH) II-1 AFAP + K Colon 52 - Mitral insufficiency - Hypertension - Hyperlipidemia Colectomy and IAA (J pouch) Negative 66 History of the proband and the family. In June 2006, at the age of 52 years, the proband (II-1) complained abdominal pain and constipation. A subsequent colonoscopy showed the presence of a rectal malignancy surrounded by numerous polyps in the rectum, sigmoid and descending colon. The patient was operated on of colectomy and ileoanal anastomosis. At anatomical examination a T3N2 (Dukes C) moderately differentiated adenocarcinoma was diagnosed; nine of the polyps (which were not counted) were analyzed and all showed features of adenomatous changes with various degrees of dysplasia. One month before colonoscopy, a chest radiogram had revealed the presence of an undefined opacity in the inferior lobe of the left lung. A subsequent CAT scan, followed by PET, confirmed the presence of a possible lung malignancy. Gastroduodenoscopy revealed chronic gastritis with intestinal metaplasia (HP-). In 2007 the patient underwent lobar resection of the left lung for removing the suspected neoplasm; the histological diagnosis was adenocarcinoma, interpreted as metastasis of colorectal cancer. The patient was treated with adjuvant chemotherapy (6 cycles). After one year (2008) the patient was again operated on for removal of a newly developed nodule at the apex of right lung. The patient is at present under chemotherapy (5-Fluorouracyl, Oxalyplatin and Folic Acid). A recent clinical evaluation revealed insufficiency of the mitral valve, hypertensive cardiomiopathy, and hyperlipidemia. Among relatives, the proband’s father died of synchronous carcinoma of the lung and prostate, at the age of 82 years. No other member of the family showed features of polyposis. Molecular characteration Genes tests negative for APC and MutYH mutations. 67 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) N. 16 1 2 I a 42 K lung 78 1 2 3 II AFAP + K colon 56 a3 1 AFAP + K colon 54 2 3 a 53 a 47 III a 59 Patient Diagnosis Age Extracolonic Manifestations Other relevant Diseases Surgery Mutations (APC/MutYH) II-2 AFAP + K Colon 56 _ Subtotal Colectomy (?) Not tested II-3 AFAP + K Colon 54 _ Abdominoperineal Resection Not tested 68 History of the proband and the family. Information about this family is scanty and fragmentary. Patient II-2 (proband’s sister) was operated on for synchronous colorectal tumors in 1973, at the age of 56 years. According to the sister, “numerous” polyps were also present, but no documentation was available. The patient died in 1990 for unknown reasons. The proband (II-3) was operated on of abdominoperineal (Miles) resection in 1987 (at the age 54 years) because of a rectal neoplasm; no polyps were referred at that time. In a subsequent endoscopic control (through the stoma), at least 20 sessile polyps were observed in the various tracts of the large bowel, ranging between 0.5 and 2.0 cm in diameter (tubular and tubulovillous adenomas with moderate-severe dysplasia). An upper endoscopy, carried out in the same year, was normal. After 3 years (1990) the patient was operated on again for a metachronous tumor of the large bowel. The patient was in relative good health in 1999; since then we lost any contact with the family. Molecular characteration No molecular test was carried out. 69 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) N. 17 1 2 I a 74 a 75 2 1 3 II K uterus 58 1 2 3 a 59 a 51 a 57 AFAP + K colon 50 a 71 4 5 a 48 a 43 6 7 a 35 a 32 III Patient Diagnosis Age Extracolonic Manifestations Other relevant Diseases Surgery II-2 AFAP + K Colon 50 Acute Pancreatitis Proctocolectomy and IAA 0 Mutations (APC/MutYH) Not tested 70 History of the proband and the family. As early as 1987, the proband (II-3) began to complain abdominal discomfort and pain. In the following years he noted frequent diarrhea with rectal bleeding and weight loss. A barium enema (1990) showed features of diffuse polyposis of the large bowel and the suspicion of an infiltrative lesion of the sigmoid. The patient was operated on of total proctocolectomy and IAA (1990). At anatomical examination, a 4 cm large TNM II (Dukes B) adenocarcinoma of the sigmoid was diagnosed, together with diffuse polyposis of all colonic segments. A total of 81 lesions were counted, ranging from a few mm up to 3.0 cm in diameter (tubular or tubulovillous adenomas with various degrees of dysplasia). During follow-up, micropolyps of the pouch and of the terminal ileum were observed and removed (1992, 1993). No extracolonic lesions could be documented. The patient died in 1997 (age 57) of acute pancreatitis. No other members of the family showed features of polyposis; in particular, all members of the third generation (including the proband’s offsprings) underwent colonoscopy, which resulted negative. It is likely therefore that the deleterious tract extinguished with the proband. Molecular characteration No test has been carried out. 71 I ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) N. 18 2 1 I K colon 48 1 K stomach 93 2 3 4 II a 77 1 a 75 a 65 2 4 5 a 63 6 7 8 3 III a 48 a 39 a 46 a2 a 39 AFAP 31 2 Patient Diagnosis Age Extracolonic Manifestations Other relevant Diseases Surgery III-1 AFAP 31 _ _ Mutations (APC/MutYH) Not tested 72 History of the proband and the family. In 2005, at the age of 31, the proband (III-8) underwent colonoscopy in absence of symptoms, but worried for polyp occurrence in the father (II-3) and one of the uncles (II-2). The examination revealed the (curious) presence of 10 polyps of large dimensions (2-3 cm in diameter) clustered in a small area of the sigmoid, at 25-30 cm from the anal verge. The remaining tracts of the bowel were normal. All polyps were removed in two successive endoscopic examinations (tubular and tubulovillous adenomas with moderate to severe dysplasia). Since then the proband has been under close surveillance, although she refused genetic testing. No surgery has been carried out. The last endoscopic control (2007) was negative. The family history is rather interesting. The paternal grandfather died of colorectal cancer at the age of 48 years (I-1); we do not know whether adenomas were presence around the malignancy. Three to 5 polyps were also removed endoscopically in the proband’s father (II-4), at the age of 52, and in the uncle (II-2), at age 67. In both cases, histology showed the coexistence of adenomatous and hyperplastic features. Molecular characteration No genetic test has been carried out. 73 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) N. 19 1 2 I AFAP + K colon 63 K colon 66 1 II 2 AFAP 35 3 AFAP 34 a 28 1 III a 10 Patient Diagnosis Age Extracolonic Manifestations Other relevant Diseases Surgery Mutations (APC/MutYH) I-2 AFAP + K Colon 63 _ Colectomy APC + II-1 AFAP 35 _ _ APC + II-2 AFAP 34 _ _ APC + 74 History of the proband and the family. The information gathered from this family is limited. The proband’s mother (I-2) was operated on of colectomy at age 63 because of a stage II (Dukes B) colorectal carcinoma over a background of polyposis (2005). In the same year the proband (II-1) underwent the first colonoscopy, which showed the presence of 20-30 polyps scattered in the various tracts of the large bowel, sessile, 4-6 mm in diameter. The removed lesions showed features of tubular adenomas with low-grade dysplasia. Subsequent endoscopies (2006 to 2008) showed a gradually increasing number of polyps, approximately of the same dimensions and histological aspect. Gastroduodenoscopy was unremarkable (2006). Thus far the patient has not been operated on. In 2006, attenuated polyposis has also been diagnosed in the proband’s sister, although further details are lacking. Genetic tests have been carried out in the proband, the proband’s mother and sister (II-2); the other sister (II-3) chose not to be tested. Molecular characteration Mutation detected Gene: APC Exon: 6 Molecular Alteration: 677-684 del 8 ins 4 (deletion of AGGACATA at position 677-684, and insertion of TTTC) Functional consequences: truncated protein. 75 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) N. 20 1 2 I a 60 a 60 1 2 II AFAP 33 a 33 1 2 III a 10 Patient Diagnosis Age Extracolonic Manifestations Other relevant Diseases Surgery II-1 AFAP 33 Gastric polyps Duodenal polyps Proctocolectomy and IAA Mutations (APC/MutYH) APC + 76 History of the proband and the family. Owing to persistent rectal bleeding, the proband (II-1) was investigated by lower endoscopy in 2000, at the age of 33 years. The endoscopist described “numerous” sessile polyps (less than 100) in the entire colon, 20 of which localized in the rectum. The removed lesions showed features of tubular and tubulovillous adenomas with moderate dysplasia. After five months, the patient was operated on of proctocolectomy and IAA. The anatomical description is lacking, however, no signs of malignancy were observed. During the hospitalization, an upper gastrointestinal endoscopy revealed the presence of numerous sessile polyps in the gastric antrum and fundus (glandular polyps), and a few sessile polyps of the duodenum (tubular adenomas with moderate dysplasia). After the operation, the patients had loosy stools and frequent defecation for several months, but the situation improved gradually over time. No other component of the family showed features of polyposis nor was affected by neoplasms. Molecular characteration Mutation detected Gene: APC Exon: 15 Type of change: 3336 del 5 Functional consequences: formation of a truncated protein. 77 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) N. 21 1 2 I a 68 1 a 74 2 3 II a 80 AFAP 75 a 81 2 3 1 III a 45 a 58 a 56 Patient Diagnosis Age Extracolonic Manifestations Other relevant Diseases Surgery II-3 AFAP 75 - Diabetes - Hypertension - Atrial fibrillation - Chronic Obstructive Lung Disease Subtotal Colectomy and IRA Mutations (APC/MutYH) In progress 78 History of the proband and the family. Starting at the age of 60 years, the proband (II-3) had a series of relevant diseases, including insulin dependent diabetes, atrial fibrillation (treated with warfarin, and digoxin), chronic obstructive lung disease (he had been a heavy smoker for many years) and hypertension (treated with diuretics and ACE-inhibitors). In June 2008 (at age 75), he noted the presence of blood in the stools; a lower endoscopy showed the presence of polyps in the large bowel. For the persistence of rectal bleeding, after a few months the patient underwent subtotal colectomy with ileorectal anastomosis. At anatomical examination, a total of 11 lesions were noted, between 1.0 and 2.0 cm in diameter, distributed both in the proximal and in the distal colon. At histology, the lesions were tubular or tubulovillous adenomas with moderate to severe dysplasia. A gastroduodenoscopy was unremarkable. Despite the numerous comorbidities, the patient recovered quite well. No other member of the family reported a clinical history of neoplasms or polyposis. Molecular characteration Search of mutations for the APC and MutYH genes in progress. 79 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) N. 22 1 2 I K colon 73 a 50 1 2 II AFAP 72 K kidney 64 2 3 1 III a 50 a 55 a 50 Patient Diagnosis Age Extracolonic Manifestations Other relevant Diseases Surgery II-2 AFAP 72 - Arterial occlusion - Chronic gastritis Subtotal colectomy and IRA Mutations (APC/MutYH) Not tested 80 History of the proband and the family. The proband, II-2, had a long history of abdominal symptoms (pain, flatulence, rectal bleeding) attributed to diverticulosis. For a further worsening of the clinical status, the patient was operated on of subtotal colectomy and ileorectal anastomosis. At anatomical examination, a total of 19 adenomatous lesions were observed scattered in the various segments of the large bowel (tubular or tubulovillous adenomas with various degrees of dysplasia), together with a few hyperplastic polyps (2006). The patient underwent a colonoscopic control in 2008, which resulted negative. Esophagogastroduodenoscopy (2006) showed chronic gastritis but no polyps. The patients had serious vascular problems, which led, few years before colectomy, to amputation of the left leg. In the family there are not other cases of polyposis. The proband’s father, however, I-1, died at the age of 73 years for on advanced colorectal malignancy. The proband’s brother died at age 64 for a neoplasm of the kidney. Molecular characteration No molecular test has been carried out. 81 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) N. 23 1 2 I K lung 73 1 a 79 2 3 II AFAP 51 a 54 1 2 3 K breast 41 4 5 III a 29 a 20 Patient Diagnosis Age II-2 AFAP 51 a 21 a 19 Extracolonic Manifestations Other relevant Diseases _ a 17 Surgery _ Mutations (APC/MutYH) Negative (MutTH) In progress (APC) 82 History of the proband and the family. Following a suggestion of the family doctor, the proband (II-2) executed a fecal occult blood test, that resulted positive. After a few months, he underwent colonoscopy (2004), which showed the presence of approximately 20 polyps distributed in the left and transverse colon. Eight of the lesions were removed for further analysis. At anatomical and histological examinations, the polyps ranged between 2.0 and 5.0 cm in diameter, with features or tubular or tubulovillous adenomas with various degrees of dysplasia; in the largest lesion a “intramucosal carcinoma” was described. At a subsequent endoscopy the remaining polyps were removed. During endoscopic follow-up (from 2005 to 2008), small sessile adenomas recurred in various segments of the large bowel (removed and examined). No other case of polyposis was reported in the family. The proband’s father presumably died of lung cancer (he was a heavy smoker). The proband’s sister was operated on for breast cancer at the age of 41 years. Molecular characteration MutYH gene analysis: negative for mutations. APC gene analysis: in progress. 83 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) N. 24 1 2 I a 64 1 KSU 84 2 II AFAP 66 a 72 1 III 2 a 40 a 38 Patient Diagnosis Age Extracolonic Manifestations Other relevant Diseases II-2 AFAP 66 Chronic Gastritis HP + Surgery _ Mutations (APC/MutYH) In progress 84 History of the proband and the family. The proband (II-2) underwent colonoscopy in 2006, at the age of 66 years, owing to a positive fecal occult blood test. The investigation showed a total of 11 polyps (from 5-6 mm to 3.0 cm in diameter) distributed in the various tracts of the large bowel. Parts of the polyps were removed, and histological examination revealed features of tubular adenomas (with moderate dysplasia) in all of the lesions. In a successive investigation, in the same year, the remaining lesions were removed and analyzed (again adenomas with various degrees of dysplasia). Newly developed adenomatous lesions were removed by lower endoscopy in 2006 and 2008. A gastroduodenoscopy (2006) showed HP + chronic gastritis of the antrum and duodenitis. No other member of the family reported polyposis. The proband’s mother died at the age of 84 for a malignancy not otherwise specified. Molecular characteration Search for mutations of the APC and MutYH genes in progress. 85 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) N. 25 1 2 3 I K colon 35 a 84 a 72 1 2 II a 63 AFAP 63 1 2 3 III a 37 a 21 Patient Diagnosis Age Extracolonic Manifestations Other relevant Diseases II-1 AFAP 63 Chronic Obstructive Lung Disease a 34 Surgery _ Mutations (APC/MutYH) In progress 86 History of the proband and the family. The proband, born in 1945, had a long history of severe chronic obstructive lung diseases, which required periods of oxygen administration especially during the night, owing to frequent episodes of nocturnal apnea; in addition, he was recently hospitalized for heart failure. He is taking several drugs, including diuretics, aspirin and corticosteroids. In 2008, because of a positive fecal occult blood test, he underwent colonoscopy, which showed the presence of at least 50 sessile polyps, 5 to 10 mm in diameter, scattered throughout the large bowel, but more dense in the proximal colon. Eight of the lesions were removed and examined (tubular adenomas, tubulovillous adenomas with moderate to high-grade dysplasia). Surgery was taken into consideration, but the decision was postponed owing to the severe lung disease. In the same year (2008), more polyps were removed at a second endoscopy. The only relevant, though not verified, information from the family was the early occurrence (35 years) of colorectal carcinoma in the proband’s paternal uncle. No other member of the family showed features of polyposis. Molecular characteration Search for constitutional mutations in the APC and MutYH genes in progress. 87 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) N. 26 1 2 I K colon 65 1 a 81 2 3 II AFAP 50 a 46 a 52 1 2 3 4 III a 23 a 19 a 6 Patient Diagnosis Age Extracolonic Manifestations Other relevant Diseases II-1 AFAP 50 - Chronic Lung Disease - Obesity - Depression - Chronic arterial occlusion - Chronic Gastritis and Duodenitis (HP+) - Hyperplastic duodenal polyps. a 22 Surgery _ Mutations (APC/MutYH) In progress 88 History of the proband and the family. The proband (II-1, born in 1957) has a long history of chronic obstructive lung disease, arterial occlusion of the legs (with claudicatio intermittens), obesity and depression, and is treated with several drugs (including antidepressant, aspirin and corticosteroids). In 2007, owing to a positive fecal occult blood test he underwent colonoscopy. This revealed the presence of nearly 30 polyps regularly distributed in the various tracts of the large bowel, ranging between 3-4 mm to 4.0 cm in diameter, both sessile and peduncolated. Eight lesions were removed and examined, and all showed features of tubular adenoma with mild to moderate dysplasia. At subsequent endoscopies (2007 and 2008) several other lesions were removed, showing histological characteristics of adenomas or hyperplastic polyps (the smaller). At the last endoscopy (February 2009), 2 small polyps were detected (and removed) in the ascending colon (hyperplastic polyps of 0.6 and 0.8 cm in diameter). A gastroduodenoscopy was carried out in January 2009; this showed severe gastritis and duodenitis (Helicobacter Pylory +) and a small (0.5 cm) hyperplastic polyp of the duodenum. In this family, no other subject showed features of polyposis; the proband’s father, however, died of colorectal cancer at the age of 65 years, but further information is lacking. Molecular characteration Search for constitutional mutations of the APC and MutYH genes in progress. 89 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) N. 27 1 2 I a 72 1 2 a 91 3 4 5 II a 74 a 71 K bladder 60 K colon 66 AFAP 67 7 1-2 III 2 3-4 a 60 8 5-6 2 2 a 37 a 35 Patient Diagnosis Age Extracolonic Manifestations Other relevant Diseases Surgery II- 4 K Colon AFAP 66 67 Obesity Hypertension Right Hemicolectomy Mutations (APC/MutYH) In progress 90 History of the proband and the family. The proband (II-4, born in 1940) is an obese individual (127 Kg, 177 cm) affected by hypertension and in treatment with several drugs (aspirin, diuretics, ACE-inhibitors, and diet). After a positive fecal occult blood test, he underwent colonoscopy (2006), which showed a vegetating lesion of the cecum. The patient was operated on of right hemicolectomy; at anatomical examination, a T2N0M0 (Dukes A) adenocarcinoma was diagnosed. Together with the malignancy, a single adenoma was noted in the resected specimen. After one year, the patient underwent a control colonoscopy (2007), which showed the presence of nearly 80 small polyps - 3 to 6 mm in diameter - evenly distributed throughout the remaining colon and rectum. Twenty of the lesions were removed and analyzed, and all showed features of adenomatous polyps with mild dysplasia. The situation was virtually unchanged at subsequent controls in September 2008 and December 2009. The patient was suggested to undergo surgical removal of the remaining colon. No other member of the family reported polyposis. The proband’s brother was operated on for a bladder malignancy at the age of 60 years. The interesting aspect of this case is the development of nearly 80 adenomas (2007) after the removal of the right colon one year before (2006), when the patient was already 67 years old. Molecular characteration Search for constitutional mutations of the APC and MutYH genes in progress. 91 ATTENUATED FAMILIAL ADENOMATOUS POLYPOSIS (AFAP) N. 28 2 1 I a 62 1 a 71 2 3 II 1 2 a 40 a 43 AFAP 35 3 5 6 4 III a 25 Patient Diagnosis a 18 a 16 a3 Age g II-1 44 AFAP Extracolonic Manifestations 1 Other relevant Diseases _ a9 a4 Surgery _ Mutations (APC/MutYH) In progress 92 History of the proband and the family. The proband (II-1) had a history of gynaecological troubles. At the age of 28 years (1988), she had the right ovary removed for a dermoid cyst; twelve years later (2000), owing to frequent vaginal bleeding and consequent anemia, she underwent extended hysterectomy. In 2003 she noticed the presence of blood in the stools, a symptom which remained overlooked for more than one year. In 2004 she executed the first lower endoscopy, which revealed the presence of “numerous” polyps (adenomas) of the large bowel. In the subsequent years she underwent several endoscopic investigations, with removal of the newly developed lesions. Despite a close control, in 2007 a total of 18 polyps were counted, 3 in the rectosigmoid, 5 in the descending and 10 in the transverse colon. All lesions showed features of tubular adenomas with low-grade dysplasia. At the last endoscopic control (2009), 14 lesions were detected (again adenomas with low-grade dysplasia), one of which was a flat adenoma of 20 mm in diameter. No other first-degree relative of the proband reported history of cancer or intestinal polyps; however, a paternal uncle died at the age of 65 years for a colorectal malignancy (not reported in the family tree). Molecular characteration Search for mutations of the APC and MutYH genes in progress. 93 SUMMARY TABLES OF THE MAIN RESULTS OBSERVED IN THE 27 FAMILIES WITH AFAP Table 1 summarizes the main clinical findings observed in 36 patients (28 families) with documented attenuated polyposis. Most of the families showed only 1 affected individual (22 out of 28, 78.6%), and in only 6 families there were 2 or more cases of AFAP. Mean age at diagnosis of AFAP was 51.8 ± 15 (mean ± SD), ranging between 18 and 75 years. Surgery was carried out in about half of the patients (18 out of 36, 50.0%); it follows that the remaining 18 individuals were managed through endoscopy. Extracolonic manifestations were identified in 5 individuals (13.9%) although in many cases they were not searched for. The number of synchronous polyps, usually detected at the first endoscopic investigation, ranged between 10 and 81, with a mean of 34.9 ± 21; in only two cases (n.15 and n.20) the investigators failed to establish the number of polyps, but preferred to say “numerous” or less than 100. In 14 out of 36 patients (39%), one or more carcinomas of the large bowel had developed at the time of diagnosis. Finally, of the 15 families in which complete genetic test could be carried out, 5 resulted positive (33.3%, 2 MutYH and 3 APC mutations). Constitutional mutations identified in 5 unrelated families are shown in details in Table 2. In family 4, the same mutation (1395delGGA of MutYH gene) was present in heterozygosis in the proband and in homozygosis in the proband’s brother. Table 3 illustrates the main surgical operations carried out in the 18 patients who underwent surgery. The data are subdivided in two study-periods (1980-1999 and 2000-2008) and by 3 different approaches. Even in the most recent times, subtotal colectomy with ileorectal anastomosis represents the most frequent type of surgery. The main clinical features of the five families which resulted positive for constitutional mutations versus the 10 families that resulted negative are illustrated in Table 4. The data are not sufficient to allow firm conclusions, however, mean age at diagnosis tends to be earlier in Mut + than in Mut – individuals; moreover, extracolonic manifestations were more frequent (in Mut + patients). Rather surprisingly, more synchronous colorectal malignancies were observed among Mut– families. Table 5 compares some of the basic clinical data between AFAP and FAP families, each subdivided in symptomatic and asymptomatic patients. Data of FAP families have already been published (1, 2, 3). It is of interest to note that FAP was diagnosed on average about 20 years earlier than AFAP, unregarding to the presence of symptoms. Moreover, in the case of AFAP there was a relatively small interval in age at diagnosis between symptomatic and asymptomatic patients. As a consequence of these findings, early diagnosis and prevention of cancer are considerably more difficult in AFAP than in FAP, as this is further demonstrated by the higher fraction of malignant neoplasms detected among asymptomatic patients (21.5% versus 9.4% in FAP). Finally, Table 6 compares constitutional mutations of the two main genes in patients with AFAP and in those with classical Adenomatosis Coli (1, 2, 3). As expected, and reported in the literature (4, 5), the percent of positivity was much lower in AFAP (33.3% vs 82.2%, though in many families molecular analysis is still in progress). While the main role played by the APC gene in FAP and quite clear, the relative contribution of APC and MutYH to the AFAP phenotype should be clarified by further investigations. 94 References 1. Ponz de Leon M, Benatti P, Percesepe A, Cacciatore A, Sassatelli R, Bertoni G, Sabadini G, Varesco L, Gismondi V, Mareni C, Montera M, Di Gregorio C, Landi P, Roncucci L. Clinical features and genotype-phenotype correlations in 41 Italian families with adenomatosis coli. Ital J Gastroenterol Hepatol. 1999 Dec;31(9):850-60. 2. Ponz de Leon M, Varesco L, Benatti P, Sassatelli R, Izzo P, Scarano MI, Rossi GB, Di Gregorio C, Gismondi V, Percesepe A, de Rosa M, Roncucci L. Phenotype-genotype correlations in an extended family with adenomatosis coli and an unusual APC gene mutation. Dis Colon Rectum. 2001 Nov;44(11):1597-604. 3. Ponz de Leon M, Pezzi A, Roncucci L, Benatti P, Rossi G, Di Gregorio C, Sassatelli R, Pedroni M, Maffei S, Borsi E, Domati F, Nozzi D, Bursi E, Mora E, De Gaetani C. Poliposi Familiare del Colon-Retto (Adenomatosis coli). L’esperienza di un Gruppo di studio sui Tumori Colorettali dell’Università di Modena e Reggio Emilia e dell’Azienda Policlinico. Università di Modena e Reggio Emilia, pag. 1-175, 2008. 4. Knudsen AL, Bisgaard ML, Bülow S. Attenuated familial adenomatous polyposis (AFAP). A review of the literature. Fam Cancer. 2003;2(1):43-55. 5. Lipton L, Tomlinson I. The genetics of FAP and FAP-like syndromes. Fam Cancer. 2006;5(3):221-6. 95 Table 1. Main clinical features in the whole study Group (28 families) Family Affected patients (n.) Age at diagnosis Sex Surgery Extracolonic manifestations N. of colorectal polyps K colon Mutation N. 1 N. 2 N. 3 N. 4 N. 5 N. 6 N. 7 N. 8 N. 9 N. 10 N. 11 N. 12 N. 13 1 1 1 2 2 1 1 2 1 1 1 1 3 65 35 49 45 50 60 59 72 18 40 62 41 44.3 + + + + + + + + + (1) + + + - 78 30 22 50 / 11 24 / 30 50 20 50 30 20 20 42 30 + + + + + + + (3) Not tested Neg Neg + (MYH) + (MYH) Neg Neg Not tested + (APC) Neg Neg Neg Neg N. 14 1 54 M F M M/F M/F F M M/F F F M F F (2)/M M - - 30 - N. 15 N. 16 N. 17 N. 18 N. 19 N. 20 N. 21 1 2 1 1 3 1 1 52 55 50 31 44.5 33 75 M F (2) M F F (3) M M + + (2) + + (1) + + + - Numerous 20 81 10 25 < 100 11 + + + + (1) - N. 22 N. 23 N. 24 1 1 1 72 51 66 M M M + - - 19 20 11 - N. 25 1 63 M - - 50 - N. 26 1 50 M - + 30 - N. 27 1 67 M + - 80 + N. 28 1 44 F - 18 - 51.8 ± 15 (mean ± SD) Range: 1875 M 19 F 17 + = 18 ─ = 18 34.9 ± 21 (mean ± SD) + = 14 ─ = 22 Patients = 36 Families = 28 +=5 In progress Neg Not tested Not tested Not tested + (APC) + (APC) In progress Not tested Neg In progress In progress In progress In progress In progress Positive = 5 Negative = 10 Not tested =6 In progress =7 96 Table 2. Constitutional mutations identified in 5 families with AFAP Family Gene Exon Type of alteration Functional Consequence Gene carriers AFAP 4 MutYH 14 1395delGGA Truncated protein 2 AFAP 5 MutYH 7 Y165C/G382D Truncated protein 2 AFAP 9 APC 15 4612delGA Truncated protein 1 AFAP 19 APC 6 677-684del8ins4 Truncated protein 3 AFAP 20 APC 15 3336del5 Truncated protein 1 Table 3. Type of colorectal surgery carried out in AFAP patients (n. 18). Period Subtotal colectomy and IRA Proctocolectomy and IAA Segmental resection or Hemicolectomy Total 1980 – 1999 4 1 1 6 2000 – 2008 6 3 3 12 97 Table 4. Clinical features in Mut + (n. 5, APC or MutYH) versus Mut – (n. 10, in which the test resulted negative) families. Mean age at diagnosis Surgery Mean number of adenomas Colorectal Cancer at diagnosis Extracolonic Manifestations Mut + (n. 5) 38.5 ± 13.0 4/5 (80%) 28.3 ± 12 2/5 (40%) 3/5 (60%) Mut – (n. 10) 49.3 ± 9.0 6/10 (60%) 28.2 ± 11 7/10 (70%) 1/10 (10%) Table 5. Age at diagnosis, number of adenomas and cancer occurrence in symptomatic and asymptomatic AFAP patients, and in patients with FAP. * Mean age at diagnosis Average number of adenomas Patients with colorectal cancer at diagnosis Symptomatic AFAP patients (n. 16) 54.9 ± 12.1 33.8 ± 22.0 8/16 (50.0%) Asymptomatic AFAP patients (n. 14) 47.9 ± 15.2 32.2 ± 19.1 3/14 (21.5%) Symptomatic FAP patients (n. 67) 35.5 ± 11.8 > 100 36/67 (53,7%) Asymptomatic FAP patients (m. 32) 23.3 ± 11.0 > 100 3/32 (9.4%) * M.Ponz de Leon et al. “Poliposi Familiare del Colon-retto”, pag 1-175; Università degli Studi di Modena e Reggio Emilia, ottobre 2008. 98 Table 6. APC and MutYH mutations detected in AFAP and FAP families. * Families (n.) Positive for mutations Total analyzed % positivity APC MutYH AFAP (28) 5 15 33.3% 3 2 FAP (61) 37 45 82.2% 33 4 * M.Ponz de Leon et al. “Poliposi Familiare del Colon-retto”, pag 1-175; Università degli Studi di Modena e Reggio Emilia, ottobre 2008. 99