Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
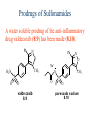
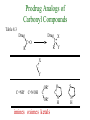
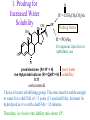
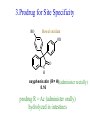
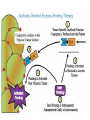
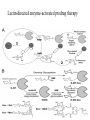
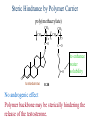
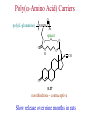
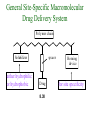
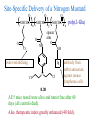
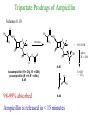
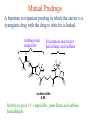
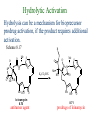
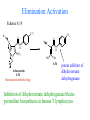
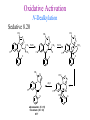
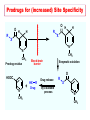
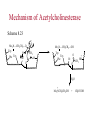
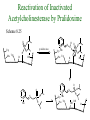
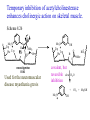
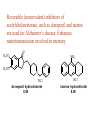
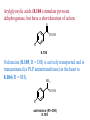
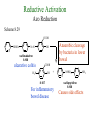
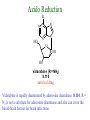
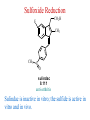
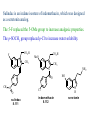
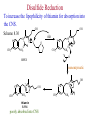
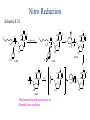
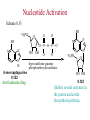
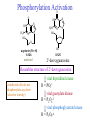
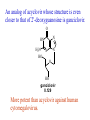
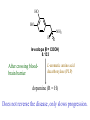
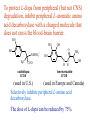
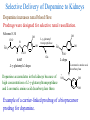
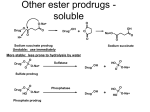
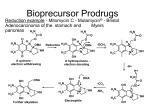
The Organic Chemistry of Drug Design and Drug Action Prodrugs and Drug Delivery Systems Nohad A Atrushi • Prodrugs and Drug Delivery Systems • Drug Discovery: 1. Drug design and development 2. Drug Design: optimizing target interaction 3. Drug Design: Optimizing access to the target Prodrugs and Drug Delivery Systems Prodrug - a pharmacologically inactive compound that is converted to an active drug by a metabolic biotransformation Ideally, conversion occurs as soon as the desired goal for designing the prodrug is achieved. Prodrugs and soft drugs are opposite: • a prodrug is inactive - requires metabolism to give active form • a soft drug is active - uses metabolism to promote excretion A pro-soft drug would require metabolism to convert it to a soft drug Utility of Prodrugs 1. Aqueous Solubility - to increase water solubility so it can be injected in a small volume 2. Absorption and Distribution - to increase lipid solubility to penetrate membranes for better absorption 3. Site Specificity - to target a particular organ or tissue if a high concentration of certain enzymes is at a particular site or append something that directs the drug to a particular site---often tried to limit the toxicity of anticancer drugs. Utility of Prodrugs (cont’d) 4. Instability - to prevent rapid metabolism; avoid firstpass effect 5. Prolonged Release - to attain a slow, steady release of the drug 6. Toxicity - to make less toxic until it reaches the site of action 7. Poor Patient Acceptability - to remove an unpleasant taste or odor; gastric irritation 8. Formulation Problems - to convert a drug that is a gas or volatile liquid into a solid Types of Prodrugs Drug Latentiation - rational prodrug design I. Carrier-linked prodrug A compound that contains an active drug linked to a carrier group that is removed enzymatically A. bipartate - comprised of one carrier attached to drug B. tripartate - carrier connected to a linker that is connected to drug C. mutual - two, usually synergistic, drugs attached to each other II. Bioprecursor prodrug A compound metabolized by molecular modification into a new compound, which is a drug or is metabolized further to a drug - not just simple cleavage of a group from the prodrug—e.g., amine getting oxidized to CO2H, to afford the active. Protecting Group Analogy for the Concept of Prodrugs Scheme 8.1 A. B. RCO2H RCH=CH2 EtOH HCl reaction on R RCO2 Et reaction on R R'CH=CH2 R'CO2Et 1 . O3 2 . H2O2 H3O+ R'CO2H R'CO2H analogous to carrier-linked The ester group can be used To modify properties of The drug analogous to bioprecursor Keep in mind that a prodrug whose design is based on rat metabolism may not be effective in humans. Mechanisms of Prodrug Activation Carrier-Linked Prodrugs Most common activation reaction is hydrolysis. Rate of hydrolysis can be modified by locating alkyl groups in area of the carbonyl group to Increase steric hindrance, and retard hydrolysis rate. Ideal Drug Carriers 1. Protect the drug until it reaches the site of action 2. Localize the drug at the site of action 3. Allow for release of drug 4. Minimize host toxicity 5. Are biodegradable, inert, and nonimmunogenic 6. Are easily prepared and inexpensive 7. Are stable in the dosage form Carrier Linkages for Various Functional Groups Alcohols, Carboxylic Acids, and Related Groups Most common prodrug form is an ester • esterases are ubiquitous • can prepare esters with any degree of hydrophilicity or lipophilicity • ester stability can be controlled by appropriate electronic and steric manipulations Drug—OH Prodrugs for AlcoholContaining Drugs Table 8.1 Ester analogs as prodrugs can affect lipophilicity or hydrophilicity Drug—OX alcoho ls X O Effect on Water Solubility C—R (R = aliphatic or aromatic) decreases (increases lipophilicity) O + C—CH 2NHMe2 increases (pKa ~ 8) O C—CH 2CH2COOO C increases (pKa ~ 5) increases (pKa ~ 4) NH + PO3= (phosphate ester) O CCH 2SO3- increases (pKa ~ 2 and ~ 6) increases (pKa ~ 1) To accelerate hydrolysis rate: • attach an electron-withdrawing group if a base hydrolysis mechanism is important • attach an electron-donating group if an acid hydolysis mechanism is important To slow down hydrolysis rate: • make sterically-hindered esters • make long-chain fatty acid esters Another Approach to Accelerate Hydrolysis Intramolecular hydrolysis of succinate esters Scheme 8.2 O -O H Drug—O- OH O Drug—OH + O - OOC Drug—O O O Also, acetals or ketals can be made for rapid hydrolysis in the acidic medium of the GI tract. COO- Enolic hydroxyl groups can be esterified as well. HOOC S F O Cl O OH N O H 2N oxind ole 8.1 antirheumatic agent O S F O Cl N O H 2N 8.2 Carboxylic Acid-Containing Groups Esterify as with alcohols Maintaining Water Solubility of Carboxylate Prodrugs O + Drug—C—O—CH2CH2—NRR'2 8.3 Can vary pKa by appropriate choice of R and R Prodrugs for Phosphate- or Phosphonate-Containing Drugs O P O O O 2 8.4 Amine Prodrugs Table 8.2 Drug—NH 2 Drug—NHX X O CR O + CCHNH3 R O O C OPh —CH 2NHCAr CHAr Amides are commonly not used because of stability Activated amides (low basicity amines or amino acids) are effective pKa of amines can be lowered by 3 units by conversion to NMannich bases (X = CH2NHCOAr) NAr N-Mannich base (R = CH2NHCOPh) has a log D7.4 two units greater than the parent compound. OH CH3 NHR ph enylpr opanolam ine h yd ro chlor ide (R =. H HCl) 8.5 Another approach to lower pKa of amines and make more lipophilic. Imine (Schiff base) prodrug OH N F Cl pr ogabid e 8.6 anticonvulsant O NH2 hydrolyze imine and amide to GABA inside brain A Reductive Carrier-Linked Prodrug Approach Scheme 8.3 OH OH NO2 Y Drug X Z 8.7 Y bio reduction Drug X N :NH Z Drug XH Y + Z 8.8 Prodrugs of Sulfonamides A water soluble prodrug of the anti-inflammatory drug valdecoxib (8.9) has been made (8.10). Ph N Ph O H 2N O CH 3 S O valde coxib 8. 9 N O Na+ N O CH3 S O O par eco xib sod ium 8.10 Prodrug Analogs of Carbonyl Compounds Table 8.3 Drug Drug X C R Y C=O R C X Y C=NR' C=NOH OR' C OR' imines oximes ketals O C N H S C N H Examples of Carrier-Linked Bipartate Prodrugs 1. Prodrug for Increased Water Solubility OH HO Me O O R' = CCH2CH2CO2 Na OR' Me O Prodrug forms R' = PO3Na2 for aqueous injection or opthalmic use R pr ed niso lone (R = R' = H) m e thylpr ednisolo ne (R = CH 3 , R' = H) 8.11 poor water solubility corticosteroid Choice of water solubilizing group: The ester must be stable enough in water for a shelf life of > 2 years (13 year half-life), but must be hydrolyzed in vivo with a half-life < 10 minutes. Therefore, in vivo/in vitro lability ratio about 106. To avoid formulation of etoposide with detergent, PEG, and EtOH (used to increase water solubility), it has been converted to the phosphate prodrug. O CH 3 O HO O O O HO O O O CH 3O OR OCH 3 etop osid e (R = H) etop osid e ph osph ate (R = PO 3 H2 ) 8. 12 2. Prodrug for Improved Absorption Through Skin HO CH3 O CH3 OR CH3 O O CH3 F O F flu ocinolo ne ace tonid e (R = H) flu ocinonide (R = COCH3 ) 8.14 corticosteroids - inflammation, allergic, pruritic skin conditions Better absorption into cornea for the treatment of glaucoma RO OH RO NHCH3 dipivefr in (R = Me 3 CCO) ep inep hr ine (R = H) 8.15 The cornea has significant esterase activity 3.Prodrug for Site Specificity RO Bowel sterilant OR N H O oxyph enis atin (R = H) (administer 8.16 prodrug R = Ac (administer orally) hydrolyzed in intestines rectally) 3. Prodrug for Site Specificity The blood-brain barrier prevents hydrophilic molecules from entering the brain, unless actively transported. The anticonvulsant drug vigabatrin (see Chapter 5) crosses poorly. A glyceryl lipid (8.17, R = linolenoyl) containing one GABA ester and one vigabatrin ester was 300 times more potent in vivo than vigabatrin. OCOR O NH2 O O NH2 O 8.17 Site Specificity Using Enzymes at the Site of Action OR RO = diethylstilbestr ol dip hosp hate (R = PO 3 ) diethylstilbestr ol (R = H) 8.18 Phosphatase should release the drug selectively in tumor cells. This approach has not been successful because the prodrugs are too polar, enzyme selectivity is not sufficient, or tumor cell perfusion rate is poor. Enzyme-Prodrug Therapies For selective activation of prodrugs in tumor cells Two steps: 1. incorporate a prodrug-activating enzyme into a target tumor cell 2. administer a nontoxic prodrug which is a substrate for the exogenous enzyme incorporated Criteria for Success with Enzyme-Prodrug Therapies 1. The prodrug-activating enzyme is either nonhuman or a human protein expressed poorly 2. The prodrug-activating enzyme must have high catalytic activity 3. The prodrug must be a good substrate for the incorporated enzyme and not for other endogenous enzymes 4. The prodrug must be able to cross tumor cell membranes 5. The prodrug should have low cytotoxicity and the drug high cytotoxicity 6. The activated drug should be highly diffusable to kill neighboring nonexpressing cells (bystander killing effect) 7. The half-life of the active drug is long enough for bystander killing effect but short enough to avoid leaking out of tumor cells Antibody-Directed Enzyme Prodrug Therapy (ADEPT) An approach for site-specific delivery of cancer drugs. Phase One: An antibody-enzyme conjugate is administered which binds to the surface of the tumor cells. The antibody used has been targeted for the particular tumor cell. The enzyme chosen for the conjugate is one that will be used to cleave the carrier group off of the prodrug administered in the next phase. Phase Two: After the antibody-enzyme has accumulated on the tumor cell and the excess conjugate is cleared from the blood and normal tissues, the prodrug is administered. The enzyme conjugated with the antibody at the tumor cell surface catalyzes the conversion of the prodrug to the drug when it reaches the tumor cell. ADEPT Advantages: 1. Increased selectivity for targeted cell 2. Each enzyme molecule converts many prodrug molecules 3. The released drug is at the site of action 4. Demonstrated to be effective at the clinical level 5. Concentrates the drug at the site of action Disadvantages: 1. Immunogenicity and rejection of antibody-enzyme conjugate 2. Complexity of the two-phase system and i.v. administration 3. Potential for leakback of the active drug An example is carboxypeptidase G2 or alkaline phosphatase linked to an antibody to activate a nitrogen mustard prodrug. I I I N N carbo xy pep tidase G2 H N O O CO2H CO2H 8.19 I + L-Glu + CO2 OH electron -do natin g; activates n itrogen mustard electron -with drawing; deactiv ates nitro gen mustard Humanization of antibodies minimizes immunogenicity. Note the prodrug-activating enzyme is a bacterial enzyme. Antibody-Directed Abzyme Prodrug Therapy (ADAPT) Instead of using a prodrug-activating enzyme, a humanized prodrug-activating catalytic antibody (abzyme) can be used. Ideally, the abzyme catalyzes a reaction not known to occur in humans, so the only site where the prodrug could be activated is at the tumor cell where the abzyme is bound. Antibody 38C2 catalyzes sequential retro-aldol and retro-Michael reactions not catalyzed by any known human enzyme. Found to be long-lived in vivo, to activate prodrugs selectively, and to kill colon and prostate cancer cells. Abzyme 38C2 Activation of a Doxorubicin Prodrug O OH O O OH O O OH OH CH3O O OH H O O CH3 NH O HO CH3O O retro-aldo l H O O 8.20 OH 3 8C2 O OH O OH H O O CH3 NH O HO O H O OH 3 8C2 OH CH3O O retro-M ichael B- OH O OH H O O CH3 NH O HO O -CO2 B- O +H+ O- OH O OH OH CH3O OH H O O CH3 O NH2 HO do xor ub icin Lectin-directed enzyme-activated prodrug therapy The LEAPT strategy. (A) Concept. LEAPT is a bipartite delivery system. (Step 1) Site-selective delivery of a glycosylated rhamnosidase (Rhacleaving) enzyme by sugar-mediated RME. (Step 2) Delivery of a Rhacapped prodrug that can be cleaved only by the delivered glycosylated rhamnosidase. When these two steps are combined, activation of the prodrug results in site-selective release of the parent drug (Step 3). (B) Glycosylated enzyme construction. Pure wild-type α-L-rhamnosidase N-WT with native “Y-linked” glycosylation was (i) chemically glycosylated with sugar-IME reagents to yield a N-WT-Xxx (Xxx = Gal, Man, or dGal) series with mixed synthetic (“X-linked”) and native (Ylinked) glycosylation or (ii) enzymatically DG with endo-H, yielding NDG. N-DG was then chemically reglycosylated with sugar-IME reagents to yield only synthetic (X-linked) N-DG-Xxx (15). An IME reagent bearing two terminal Gal units (dGal) was also synthesized from Dgalactose. Lectin-directed enzyme-activated prodrug therapy Gene-Directed Enzyme Prodrug Therapy (GDEPT) Also known as suicide gene therapy A gene encoding the prodrug-activating enzyme is expressed in target cancer cells under the control of tumor-selective promoters or by viral transfection. These cells activate the prodrug as in ADEPT. Cl Cl N O2N N O NMe O OMe OMe NH O N H OMe OMe n it roreduct ase N H HO N: O NMe N O NH N H OMe OMe -CO 2 +H+ O Cl OMe N O Scheme 8.6 N H OMe OMe NH2 am ino -seco-CBI -TMI 8.21 Nitroreductase gene-directed enzyme prodrug therapy suicide gene therapy suicide gene therapy 4.Prodrug for Stability protection from first-pass effect Oral administration has lower bioavailability than i.v. injection. O NHCH(CH3 )2 OR' R pr op ano lol (R = R' = H) 8.22 antihypertension O pr od ru g R' = CCH2 CH2COOH plasma levels 8 times that with propanolol 5.Prodrugs for Slow and Prolonged Release 1. To reduce the number and frequency of doses 2. To eliminate night time administration 3. To minimize patient noncompliance 4. To eliminate peaks and valleys of fast release (relieve strain on cells) 5. To reduce toxic levels 6. To reduce GI side effects Long-chain fatty acid esters hydrolyze slowly Intramuscular injection is used also O F N OR Cl halop er idol (R = H) halop er idol decan oate (R = CO(CH 2) 8CH3 ) 8.24 Sedative/tranquilizer/antipsychotic O prod ru g R' = C(CH2)8CH3 slow release inject i.m. Antipsychotic activity for about 1 month 6.Prodrugs to Minimize Toxicity Many of the prodrugs just discussed also have lowered toxicity. For example, epinephrine (for glaucoma) has ocular and systemic side effects not found in dipivaloylepinephrine. 7.Prodrug to Increase Patient Acceptance The antibacterial drug clindamycin (8.28) is bitter and not well tolerated by children. CH3 N H H N H O HO Cl O OH SCH3 OR Clindamycin palmitate clind om ycin (R = H) clind om ycin pho sphate (R = PO is not bitter. 3H 2) clind om ycin palm itate (R = O(CH 2) 14 CH3 ) 8.28 Either not soluble in saliva or does not bind to the bitter taste receptor or both. 8.Prodrug to Eliminate Formulation Problems Formaldehyde is a gas with a pungent odor that is used as a disinfectant. Too toxic for direct use. N CH2O + NH3 H+ H3O+ N N N me thenamin e 8.30 It is a stable solid that decomposes in aqueous acid. The pH of urine in the bladder is about 4.8, so methenamine is used as a urinary tract antiseptic. Has to be enteric coated to prevent hydrolysis in the stomach. Areas of Improvement for Prodrugs Areas of Improvement for Prodrugs • site specificity • protection of drug from biodegradation • minimization of side effects Macromolecular Drug Delivery To address these shortcomings, macromolecular drug delivery systems have been developed. A bipartate carrier-linked prodrug in which the drug is attached to a macromolecule, such as a synthetic polymer, protein, lectin, antibody, cell, etc. Absorption/distribution depends on the physicochemical properties of macromolecular carrier, not of the drug. Therefore, attain better targeting. Minimize interactions with other tissues or enzymes. Fewer metabolic problems; increased therapeutic index. Disadvantages of Macromolecular Delivery Systems • Macromolecules may not be well absorbed • Alternative means of administration may be needed (injection) • Immunogenicity problems Macromolecular Drug Carriers Synthetic polymers CH2 CH CH2 x OH CH y O O O 8.32 O Aspirin linked to poly(vinyl alcohol) has about the same potency as aspirin, but less toxic. Steric Hindrance by Polymer Carrier poly(methacrylate) CH3 CH2 C x C=O O CH3 CH2 C y C=O O S=O O testosterone to enhance water solubility 8.34 No androgenic effect Polymer backbone may be sterically hindering the release of the testosterone. A spacer arm was added, and it was as effective as testosterone. CH3 CH3 CH2 C x CH2 C y C=O C=O O sp acer arm O + Cl NMe2 - O O O 8.35 S=O H3 C to enhance water solubility Poly(-Amino Acid) Carriers poly(L-glutamine) O NH CH C x spacer O N H O O O C CH O 8.37 norethindrone - contraceptive Slow release over nine months in rats General Site-Specific Macromolecular Drug Delivery System Po ly mer ch ain Solubilizer either hydrophilic or hydrophobic sp acer Drug 8.38 Ho ming dev ice for site specificity Site-Specific Delivery of a Nitrogen Mustard O NHCH C O x O NHCH C NH poly(L-Glu) O NH water-solubilizing Ig Cl z spacer arm O O- y O NHCH C N Cl antibody from rabbit antiserum against mouse lymphoma cells 8.39 All 5 mice tested were alive and tumor free after 60 days (all controls died). Also, therapeutic index greatly enhanced (40 fold). Tumor Cell Selectivity Drug attached to albumin (R = albumin) Tumor cells take up proteins rapidly. Proteins broken down inside cells, releasing the drug. Shown to inhibit growth of Ectomelia virus in mouse liver, whereas free inhibitor did not. NH2 N RO O N O HO OH cytosine arabinoside (R = H) 8.41 antitumor Antibody-Targeted Chemotherapy Scheme 8.7 calicheamicin, except as disulfide instead of trisulfide HO O O NH CH3O H HO polysaccharide O CH3 antibo dy Lys S O O O humanized H N N H N O S RS - O NH CH3O H O S ge m tuz um ab oz ogam icin 8.42 spacer Does not release calicheamicin nonenzymatically. Exhibits no immune response. 8.43 polysaccharide Tripartate Drugs (Self-immolative Prodrugs) A bipartate prodrug may be ineffective because the linkage is too labile or too stable. In a tripartate prodrug, the carrier is not attached to the drug; rather, to the linker. Therefore, more flexibility in the types of functional groups and linkages that can be used, and it moves the cleavage site away from the carrier. The linker-drug bond must cleave spontaneously (i.e., be self-immolative) after the carrier-linker bond is broken. Tripartate Prodrugs Scheme 8.8 Carrier Link er Drug enzy me Carrier + Link er Drug sp on tan eous Link er + Drug Typical Approach Scheme 8.9 O Drug—X—CH2 —O— CR esterase Drug—X—CH2 —O- + fast Drug—X- + CH2O RCOOH Tripartate Prodrugs of Ampicillin O Poor oral absorption (40%) Excess antibiotic may destroy important intestinal bacteria used in digestion and for biosynthesis of cofactors. Also, more rapid onset of resistance. NH S NH2 O N COO- am picillin 8.44 antibacterial Various esters made were too stable in humans (although they were hydrolyzed in rodents) - thought the thiazolidine ring sterically hindered the esterase. Tripartate Prodrugs of Ampicillin Scheme 8.10 O Ph O Ph NH S NH2 O esterase N O O O R R' NH S NH2 O + N .. OH O O O R'COOH wh en R' = OEt R 8.46 bacam picillin (R = CH3 , R' = OEt) pivam picillin (R = H, R' =t-Bu ) 8.45 98-99% absorbed EtOH + CO2 O R 8.44 Ampicillin is released in < 15 minutes H Reversible Redox Drug Delivery System to the CNS (Nitrogen Mustard?????HW) Scheme 8.11 hydrolysis activated hydrolysis deactivated O O Drug—X N CH3 crosses blo od-brain barrier O Drug—X N CH3 enzy me o xidatio n 8.47 hydrophilic drug HOOC electron-donating, lipophilic carrier + N+ CH3 8.49 Drug — XH Drug—X 8.48 N+ CH3 enzy me h ydrolysis electronwithdrawing; hydrophilic Passive diffusion of 8.47 into the brain; active transport of 8.49 out of the brain XH of the drug is NH2, OH, or COOH If oxidation occurs before it gets into the brain, it cannot cross the blood-brain barrier. When the drug is a carboxylic acid, a selfimmolative reaction also can be used. Scheme 8.12 O O Drug C OCH2 8.50 O C carrier esterase O O Drug—C— O—CH2 —O- + HOC fast —CH2O Drug—COO- carrier Example of Redox Drug Delivery Antibody generation in the brain is not significant. -Lactams are too hydrophilic to cross the blood-brain barrier effectively. H N R O O S O N O enzy matic O o xidatio n O O H N R S O O O O N Me 8.51 O N N Me esterase H N R O Scheme 8.13 O S H N R -CH 2O N O- O O O S N O High concentrations of -lactams delivered into brain. O O O O N Me 8.49 Tripartate Prodrug for Delivery of Antibacterials Permeases are bacterial transport proteins for uptake of peptides. O Scheme 8.14 HN + H3N O O CH3 O F N N H + H3N 1 . p ermease COO - 2 . p ep tidase COO- O + CH3 .. H2N O 8.53 + H2N + NH4 + O H COO H COO- Only L,L-dipeptides are active F HN - + F HN O N H N COO- Mutual Prodrugs A bipartate or tripartate prodrug in which the carrier is a synergistic drug with the drug to which it is linked. Antibacterial ampicillin -lactamase inactivator penicillanic acid sulfone O Ph NH2 N H O O S N O S O O O N O O su ltam icillin 8.59 Hydrolysis gives 1:1:1 ampicillin : penicillanic acid sulfone : formaldehyde Ideal Mutual Prodrugs • Well absorbed • Both components are released together and quantitatively after absorption • Maximal effect of the combination of the two drugs occurs at 1:1 ratio • Distribution/elimination of components are similar II.Bioprecursor Prodrugs Carrier-linked prodrugs largely use hydrolytic activation Bioprecursor drugs mostly use oxidative or reductive activation The metabolically-activated alkylating agents are actually examples of bioprecursor prodrugs. Protonation Activation Discovery of Omeprazole Cimetidine and ranitidine reduce gastric acid secretion by antagonizing the H2 histamine receptor. Another way to lower gastric acid secretion is by inhibition of the enzyme H+,K+-ATPase (also called the proton pump), which exchanges protons for potassium ions in parietal stomach cells, thereby increasing stomach acidity. Lead Discovery Lead compound found in a random screen. S NH2 N 8.60 Liver toxicity observed thought to be because of the thioamide group. Lead Modification Related analogs made, and 8.61 had good antisecretory activity. N S N 8. 61 N H Modification gave 8.62 with high activity. S N 8.62 N N H The sulfoxide (8.63) was more potent, but it blocked iodine uptake into the thyroid. O S N N N H tim opr az ole 8.63 Lead Modification (cont’d) Modification of 8.63 gave 8.64 having no iodine blockage activity. CH3 N Picoprazole shown to be an inhibitor of H+,K+-ATPase. SAR of analogs indicated electron-donating groups on H3 C the pyridine ring were favorable. Increased inhibition of H+,K+-ATPase. Best analog was omeprazole (8.65). O S CO2CH3 N N H picop razo le 8.64 OCH3 CH3 N O S CH3 N N H om epr az ole 8.65 OCH3 The pKa of the pyridine ring of omeprazole is about 4, so it is not protonated and able to cross the secretory canaliculus of the parietal cell. The pH inside the cell is below 1, so this initiates the protonation reaction below. Scheme 8.16 OCH3 CH3 H3 C OCH3 CH3 H3 C N .. OCH3 H3 C N S HN N O S H+ OCH3 OCH3 O .. HN S N H OCH3 CH3 H3 C -H2 O N HN N CH3 N OH S S N N OCH3 OCH3 8.66 om epr az ole 8.65 Formation of 8.66 leads to covalent attachment. Omeprazole also inhibits isozymes of carbonic anhydrase, another mechanism for lowering gastric acid secretion. +H+ OCH3 CH3 H3 C N N S NH OCH3 S Hydrolytic Activation Hydrolysis can be a mechanism for bioprecursor prodrug activation, if the product requires additional activation. Scheme 8.17 S OS+ Me O N H R S S O Me O- S S+ N N K2CO3/RX N Me HO O O Me OH Me HO leinam ycin 8.70 O antitumor agent Me HO 8.71 O prodrugs of leinamycin HO- Hydrolysis of these analogs gives an intermediate that reacts further to the activated form. O O O Me S O O Me - S+ N S O N Me HO S- S+ N O O O S N Me HO O Me Me O- O Me S OS+ Me O S N H N O Me OH DNA cleavage Me O O O O this was synthesized and gave 8.74 as well as DNA cleavage O O 8.73 8.72 increased stability over leinamycin HO2 C Me HO Scheme 8.18 Me O N H S N S HO Me O O 8.74 O Elimination Activation Scheme 8.19 B: CF 3 O H N N H N O B CH3 H le fluno m ide 8.75 rheumatoid arthritis drug CF 3 O C HO N H CH3 8.76 potent inhibitor of dihydroorotate dehydrogenase Inhibition of dihydroorotate dehydrogenase blocks pyrimidine biosynthesis in human T lymphocytes. Oxidative Activation N-Dealkylation Sedative 8.20 CH3 N N N Cl N O CH3 CH3 CH3 N N CH3 P4 50 N O Cl X CH3 P4 50 Cl X CH3 N Cl X alpr az oalam (X = H) tr iazolam (X = Cl) 8.77 N N N -H2 O N NH .. 2 O H N N N N X CH3 N N .. NH Cl HO X O-Dealkylation Analgesic activity of phenacetin is a result of O-dealkylation to acetaminophen. O HN CH 3 OR phe nacetin (R = CH2 CH3 ) acetam ino phen (R = H) 8.78 Oxidative Deamination Neoplastic (cancer) cells have a high concentration of phosphoramidases, so hundreds of phosphamide analogs of nitrogen mustards were made for selective activation in these cells. HO: H N P P-4 50 O Scheme 8.21 P Cl O N + NH3 Cyclophosphamide was very effective, but it required liver homogenates (contains P450) for activation. Therefore oxidation is required, not hydrolysis. H NH 2 O Cl O N Cl cycloph osph am ide 8.79 HPO4= O H N H O 8.80 Cl O Cl DNA H + Cl 8.83 8.82 8.84 DNA O + N HN H 2N Cl 8.81 H 2N O -O P spo ntan eo us N or p ho sph oramidase Cl Cl B: Cl + HN P O N N N DNA H N H N O OH N HN H 2N N N 8.86 8.85 isolated OH N-Oxidation identified O CH3 NHNHCH2 CNHCHMe 2 O O P4 50 CH3 NHNHCH2 CNHCHMe 2 -H2 O HO pr ocar bazine 8.87 CH3N=N—CH + B— H H CNHCHMe 2 8.88 B: advanced Hodgkin’s disease O CH3N-NH2 + OHC H O H 2O CNHCHMe 2 CH3N-N=CH H CNHCHMe 2 8.89 CH3N=NH O + CH3-N CH3 Scheme 8.22 N + N2 DNA HN H 2N N CH3 +N N DNA O CH3 N N N HN H2N 8.90 identified N-Oxidation Pralidoxime chloride is an antidote for nerve poisons. It reacts with acetylcholinesterase that has been inactivated by organophosphorus toxins. +N Cl CH3 - N OH pr alido xim e chlor id e 8.91 To increase the permeability of pralidoxime into the CNS, the pyridinium ring was reduced (8.92). N CH 3 8.92 N OH oxidation into brain Cl - +N N OH CH3 pr alido xim e chlor id e 8.91 Similar to the reversible redox drug delivery strategy for getting drugs into the brain by attaching them to a dihydronicotinic acid, hydrophobic 8.92 crosses the blood-brain barrier; oxidation to 8.91 prevents efflux from brain. Prodrugs for (increased) Site Specificity ✵Bodor and co-workers have devised a reversible redox drug delivery system (RRDDS) for getting drugs into the CNS and then, once in, preventing their efflux. The approach is based on the attachment of a hydrophilic drug to a lipophilic carrier (a dihydropyridine) thereby making prodrug that actively transported into the brain. O H R H R X Prodrug residue H X N N CH3 O H CH3 Blood-brain barrier Prodrugs for (increased) Site Specificity Once inside the brain, the lipophilic carrier is converted enzymatically to a highly hydrophilic species (positively charged), which is then enzymatically hydrolyzed back to the drug and N-methylnicotinic acid, which is eliminated from the brain. O H R H X N ♫The oxidation of the dihydropyridine to the CH3 Enzymatic oxidation pyridinium ion (half-life generally 20-50 min) prevents the drug from escaping out of the brain because it becomes charged. ♫This drives the equilibrium of the lipophilic precursor throughout all of the tissues of the body to favor the brain. O R X N CH3 Prodrugs for (increased) Site Specificity O H R H R X O H H X N N CH3 Prodrug residue CH3 Blood-brain barrier Enzymatic oxidation O HOOC + N CH3 HX Drug R Drug release by a suitable process R X N CH3 Mechanism of Acetylcholinesterase Scheme 8.23 O Me 3N—CH2CH2—O Trp H CH3 Trp Phe B+ :B H O Me 3N—CH2CH2—OH Trp Trp Phe O B: O CH3 + HB H 2O + M e3NCH2CH2OH + CH3 COOH Inactivation of Acetylcholinesterase by Diisopropyl Phosphorofluoridate Scheme 8.24 affinity labeling agent O Trp Phe Trp O O F P O H B+ O H 8.93 P Trp :B Phe Trp O B O O HB irreversible inhibition Inactivation prevents degradation of the excitatory neurotransmitter acetylcholine. Accumulation of acetylcholine causes muscle cells in airways to contract and secrete mucous, then muscles become paralyzed. Reactivation of Inactivated Acetylcholinesterase by Pralidoxime Scheme 8.25 O P Trp Phe Trp O B O p ralidox ime O HB O N Me N O P O O N Trp Me Phe Trp N Trp Me Phe Trp N H O O: B O P O O HB O N P O O O BH OH :B Temporary inhibition of acetylcholinesterase enhances cholinergic action on skeletal muscle. Scheme 8.26 O Me 3N Trp Phe Trp O H B+ O NMe2 :B H ne ostig m ine 8.94 Used for the neuromuscular disease myasthenia gravis Me 3N Trp Trp Phe OH B: NMe2 O covalent, but reversible slo w inhibition H2O + Me 3N OH + HB O CO2 + Me2NH Reversible (noncovalent) inhibitors of acetylcholinesterase, such as donepezil and tacrine are used for Alzheimer’s disease. Enhances neurotransmission involved in memory. H 3CO H 3CO O NH 2 N . HCl do nepe zil hydr och lor ide 8.95 N . HCl tacr ine hydr och lor ide 8.96 S-Oxidation Poor oral bioavailability of brefeldin A. Converted to Michael addition sulfide prodrug (8.98). S-Oxidation and elimination gives brefeldin A. Scheme 8.27 H HO O HO O H :B H HO CH3 RSH HO RS H H brefeldin A 8.97 O O [O] CH3 HO H 8.98 -RSOH antitumor, antiviral agent H HO RS O H H O H 8.99 O CH3 Aromatic Hydroxylation Cyclohexenones as prodrugs for catechols Scheme 8.28 N N P4 50 N N -H2 O HO O O 8.100 oxidation next to sp2 carbonyl OH O aromatic hydroxylation P4 50 N HO OH 8.101 Alkene Epoxidation O N N O NH2 car bam az epine 8.102 O NH2 8.103 active anticonvulsant agent Transamination Stimulation of pyruvate dehydrogenase results in a change of myocardial metabolism from fatty acid to glucose utilization. Glucose metabolism requires less O2 consumption. Therefore, utilization of glucose metabolism would be beneficial to patients with ischemic heart disease (arterial blood flow blocked; less O2 available). Arylglyoxylic acids (8.104) stimulate pyruvate dehydrogenase, but have a short duration of action. O COOH R 8.104 Oxfenicine (8.105, R = OH) is actively transported and is transaminated (a PLP aminotransferase) in the heart to 8.104 (R = OH). NH2 COOH R oxfenicin e (R = OH) 8.105 Reductive Activation Azo Reduction Scheme 8.29 COOH N NHSO 2 N=N Anaerobic cleavage by bacteria in lower bowel OH su lfas alazi ne 8.106 ulcerative colitis COOH H 2N OH + 8.107 For inflammatory bowel disease N NHSO2 NH2 su lfap yr idine 8.108 Causes side effects To prevent side effect by sulfapyridine a macromolecular delivery system was developed. poly(vinylamine) n Not absorbed or metabolized in small intestine. NH SO2 spacer CO2Na N N OH 8.109 CO 2Na NH 2 OH Released by reduction at the disease site. Sulfapyridine is not released (still attached to polymer). More potent than sulfasalazine. Azido Reduction R N N HO N N O OH HO vidarabin e (R = NH2) 8.110 antiviral drug Vidarabine is rapidly deaminated by adenosine deaminase. 8.110, R = N3 is not a substrate for adenosine deaminase and also can cross the blood-brain barrier for brain infections. Sulfoxide Reduction CO 2H F CH 3 CH 3 S O sul indac 8. 111 anti-arthritis Sulindac is inactive in vitro; the sulfide is active in vitro and in vivo. Sulindac is an indane isostere of indomethacin, which was designed as a serotonin analog. The 5-F replaced the 5-OMe group to increase analgesic properties. The p-SOCH3 group replaced p-Cl to increase water solubility. CO 2H F CO2H MeO CH 3 CH3 N O CH 3 S O sul indac 8. 111 Cl in dome thacin 8.112 NH2 HO N H se ro tonin Disulfide Reduction To increase the lipophilicity of thiamin for absorption into the CNS. OH Scheme 8.30 CH 3 .. GSH N N S S NH2 N N O H O N OH N CH 3 O H NH 2 +B 8.113 S- H nonenzymatic OH + N N CH 3 N NH 2 S thiam in 8.114 poorly absorbed into CNS N: N OH CH 3 N S NH 2 OH CH3O N HN NH2 To diminish toxicity of primaquine and target it for cells with the malaria parasite, a macromolecular drug delivery system was designed. pr im aqu ine 8.115 Intracellular thiol much higher than in blood; selective reduction inside the cell antimalarial, toxic CH3O lactose N HN primaquine O N H S S NH3+ 8.116 serum albumin for improved uptake in liver lactose Therapeutic index of 8.116 is 12 times higher than 8.115 in mice. Nitro Reduction Scheme 8.32 O HN O Cl N H3 C P O N Cl bio reduction N H3 C HO P O O O -O P O- O O O O N O O- O H 2O P O O HO 8.121 Mechanism-based inactivator of thymidylate synthase HO N H3 C P F HN O N O O O F HN O N O 8.119 F O P F HN O HO 8.120 O HN H3 C HO .. HO NH O Cl .. N N O O 8.118 F HN O O O O O 2N O O F O O N O OHO Nucleotide Activation Scheme 8.33 = O3PO O SH N N N N H 6-mer captop ur ine 8.122 Anti-leukemia drug O SH O O—P—O—P—OHO OH O - O - N N = O3PO N N O h yp ox an thine-guan ine p ho sph oribo syltran sferase HO OH 8.123 Inhibits several enzymes in the purine nucleotide biosynthesis pathway. Phosphorylation Activation O O N HN H2N RO N N N HN H 2N HO N N O O acyclo vir (R = H) 8.124 antiviral HO 8.125 2-deoxyguanosine Resembles structure of 2-deoxyguanosine Uninfected cells do not phosphorylate acyclovir (selective toxicity) viral thymidine kinase R = PO3= viral guanylate kinase R = P2O6-3 viral phosphoglycerate kinase R = P3O9-4 Acyclovir triphosphate is a substrate for viral -DNA polymerase but not for normal -DNA polymerase Incorporation into viral DNA leads to a dead-end complex (not active). Disrupts viral replication cycle and destroys the virus. Even if the triphosphate of acyclovir were released, it is too polar to be taken up by normal cells. High selective toxicity Resistance to Acyclovir Modification of thymidine kinase Change in substrate specificity for thymidine kinase Altered viral -DNA polymerase Only 15-20% of acyclovir is absorbed Therefore, prodrugs have been designed to increase oral absorption. Hydrolyzes here NH2 N N H 2N N N Prodrug for a prodrug HO O 8.126 adenosine deaminase acyclovir Hydroxylates here N N H 2N HO N N O 8.127 xanthine oxidase acyclovir This prodrug is 18 times more water soluble than acyclovir. A bipartate carrier-linked prodrug of acyclovir, the L-valyl ester of acyclovir, has 3-5 fold higher oral bioavailability with the same safety profile. O N HN H2N H2N O N N O O valaciclo vir 8.128 An analog of acyclovir whose structure is even closer to that of 2-deoxyguanosine is ganciclovir. O N HN H 2N HO N N O HO ganciclovir 8.129 More potent than acyclovir against human cytomegalovirus. Two carbon isosteres of ganciclovir are available. O N HN H 2N HO N H 2N AcO N HO pen ciclovir 8. 130 N N C in place of O N N AcO C in place of O fam ciclo vir 8.131 Better oral absorption than penciclovir Converted to penciclovir rapidly Sulfation Activation Activity of minoxidil requires sulfotransferase-catalyzed sulfation to minoxidil sulfate. OR H 2N N NH2 N N minoxidil (R = ) minoxidil sulfate (R = SO 8.132 3 - ) hair growth Inhibitors of the sulfotransferase inhibit the activity of minoxidil, but not minoxidil sulfate. Decarboxylation Activation An imbalance in the inhibitory neurotransmitter dopamine and the excitatory neurotransmitter acetylcholine produces movement disorders, e.g. Parkinson’s disease. In Parkinson’s there is a loss of dopaminergic neurons and a low dopamine concentration. Dopamine treatment does not work because it cannot cross blood-brain barrier, but there is an active transport system for L-dopa (levodopa, 8.133, R = COOH). HO HO NH 2 H R levodo pa (R = COOH) 8.133 After crossing bloodbrain barrier L-aromatic amino acid decarboxylase (PLP) dopamine (R = H) Does not reverse the disease, only slows progression. Combination Therapy Monoamine oxidase B (MAO B) degrades dopamine in the brain. Therefore, a MAO B-selective inactivator is used to protect the dopamine - selegiline (L-deprenyl). Peripheral L-aromatic amino acid decarboxylase destroys >95% of the L-dopa in the first pass. Maybe only 1% actually gets into brain. To protect L-dopa from peripheral (but not CNS) degradation, inhibit peripheral L-aromatic amino acid decarboxylase with a charged molecule that does not cross the blood-brain barrier. HO HO HO NHNH 3 H3 C COO - car bid opa 8.134 + OH O HO N N H H NH2 OH be nser azid e 8.135 (used in U.S.) (used in Europe and Canada) Selectively inhibits peripheral L-amino acid decarboxylase. The dose of L-dopa can be reduced by 75%. Selective Inactivation of Brain Monoamine Oxidase A Earlier we found that inactivation of brain MAO A has an antidepressant effect, but a cardiovascular side effect occurs from inactivation of peripheral MAO A. 8.136 (R = COOH) is actively transported into the brain, where L-aromatic amino acid decarboxylase converts it to 8.136 (R = H), a selective MAO A mechanism-based inactivator. Carbidopa protects 8.136 (R = COOH) from decarboxylation outside of the brain. F NH 2 R OH 8.136 Selective Delivery of Dopamine to Kidneys Dopamine increases renal blood flow. Prodrugs were designed for selective renal vasodilation. Scheme 8.34 COO OH - H N + NH3 O OH COO- L--glutamy l transp eptidase OH + NH3 Glu 8.137 OH COO- L-dopa L-aromatic amino acid decarbox ylase L--glutamyl-L-dopa Dopamine accumulates in the kidneys because of high concentrations of L--glutamyltranspeptidase and L-aromatic amino acid decarboxylase there. + NH3 OH OH Example of a carrier-linked prodrug of a bioprecursor prodrug for dopamine.