Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Embryonic stem cell wikipedia , lookup
Biochemistry wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
Cell culture wikipedia , lookup
Homeostasis wikipedia , lookup
Organisms at high altitude wikipedia , lookup
Neuronal lineage marker wikipedia , lookup
Hematopoietic stem cell transplantation wikipedia , lookup
Adoptive cell transfer wikipedia , lookup
Hematopoietic stem cell wikipedia , lookup
State switching wikipedia , lookup
Artificial cell wikipedia , lookup
Cell theory wikipedia , lookup
Organ-on-a-chip wikipedia , lookup
Developmental biology wikipedia , lookup
Evolution of metal ions in biological systems wikipedia , lookup
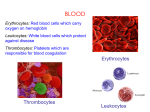
Lecture 5 Erythropoiesis Anemia: a reduced carrying capacity of oxygen. Anemia is not a reduced hematocrit (Hct), because you can have less O2 for other reasons. You can be anemic with normal levels of RBC’s if the hemoglobin is abnormal, and has less iron. Blood transports O2, CO2, nutrients, wastes, hormones, lipids, etc. The blood also regulates body temperature. Hormones can adjust blood volume, urine output, and maintain pH (which needs to be 7.40). A person with blood pH of 7.35 or 7.45 will not feel well, act strangely. There are buffers in the blood to keep the pH steady, and lungs and kidneys help with this. We also have WBCs in our blood for immune system function, and platelets for clotting. Adults have 4-6 liters of blood (Let’s call it 5 liters for this class). The heart pumps all of it every 60 seconds. The temperature of our core is higher than the superficial temperature. Blood is 5x thicker than water because of the RBC’s. Increase the numbers of RBC’s, and you will increase viscosity, increase heart workload. The color of blood is bright red or dark burgundy, depending on oxygenation. Blood is intracellular fluid (RBC’s, 45%) and extra cellular fluid (plasma, 55%). In plasma is mostly water, plus dissolved hormones, proteins, electrolytes. Don’t memorize the slide that shows everything in the blood; just understand that there are many things in the blood. Water is the solvent. 7g% of the plasma is protein, albumin is the most abundant. Albumin helps keep the water in the plasma by keeping the particle count high. Problems with water leaving the vascular compartment will lead to ascites, so albumin in the blood acts as osmotic support. There are other proteins, such as fibrinogen (inactive form of fibrin). Fibrin is the fiber that grows across a cut and makes a scab. Other proteins include regulatory and proteolytic enzymes. Other substances in blood include glucose, lipids, cholesterol, bilirubin (waste product of RBC destruction), and creatinine (waste product of kidney). We check those levels in the blood to ascertain kidney function. There are lots of solutes in the blood! BLOOD ANALYSIS TERMINOLOGY You need to measure hematocrit (Hct), hemoglobin (Hgb), and do a RBC count. With this information, you can then calculate MCV, MCH, and MCHC. By the way, RBC = corpuscle. MCV(mean corpuscular volume) calculated as MCV = Hct / RBC count. Normal is 80-100 µm MCH (mean corpuscular hemoglobin) calculated as MCH = Hb / RBC count. Normal is 32 pcicograms (10 -12 micrograms). There is not much, but hemoglobin is still the most abundant protein in RBC’s. MCHC (mean corpuscular hemoglobin concentration) is the ratio of MCH to MCV. It is calculated as MCHC = Hb/Hct. Normal is 34% MCD (mean corpuscular diameter) normal is 7-8 µm wide, 2 µm thick. COLOR (Chromasia; indicates how much Hgb is there) Normal is 1.0 Normochromic is 1 Hypochromic is less than 1. Can’t have hyperchromic RBC’s. A disorder that appears hyperchromic is Hereditary Spherocytosis. It is the most common inherited disorder that affects the RBC membrane. RBC comes out of marrow normal, but with time, the membrane is lost, but none of the contents are lost, RBC gets smaller and concentrated, looks redder. Thus, it could look hyperchromatic. SIZE Normocytic is normal size. Microcytic (small) Macrocytic or Megaloblastic (big) SHAPE Poikilocytosis is abnormal shapes, such as sickled or sphaerocytes. Cells with abnormal size or shape will be removed from circulation faster, get anemia. CELL COUNTS We have 5 liters of blood, 25 trillion RBC’s, 4.5 million in one microliter. We need the rate of old blood cells that die to match the rate of new ones being made. RBC’s are made by stem cells in bone marrow. Too many RBC’s is polycythemia, too few is anemia. RBC shape should be biconcave because that increases surface area for O2 binding and allows it to be flexible to get through capillaries. When a RBC gets older, its membrane is less flexible, gets stuck in liver and is detected and destroyed. Because of the shape increasing surface area, there is rapid gas exchange across cell membranes for wastes and O2. You know that hemoglobin binds O2, but CO2 can also bind to hemoglobin, but only if O2 is not there. SITES OF HEMOPOIETIC ACTIVITY An embryo (< 3 months) makes blood in yolk sac Fetus (> 3 months) makes blood in the liver At birth, makes blood in the bone marrow Blood that is made in the bone marrow is specifically made in the axial and appendicular skeleton until the age of 20. Thereafter, it can only be made in the axial skeleton, mainly in the sternum and iliac crest. ERYTHROPOIESIS (MAKING A RBC) A stem cell that is completely undifferentiated yet is called a pleuripotent stem cell. A pleuripotent stem cell can differentiate into any cell type: nerve, muscle, blood, etc. Since we are discussing blood cell formation, we will focus on a pleuripotent stem cell that differentiates into a Hemocytoblast. Hemocytoblasts can differentiate into any blood cell type. Therefore, they are not pleuripotent anymore, but they are still multipotent (they are “determined”, but not completely differentiated). A hemocytoblast will continue to differentiate into one of two cell types: 1. Lymphoid line generates B and T cells 2. Myeloid line generates red and all other blood cells. Scientists doing stem cell research have found a way to turn a multipotent cell back into a pleuripotent cell by just Inserting four genes into a multipotent cell! To make a good RBC, you need to start with good ingredients: a good hemocytoblast, proper nucleotides, folic acid, vitamin B12, and other vitamins. You also need growth inducers, differentiation markers (signals), amino acids (Adenine, Thymine, cytosine, guanine), heme (a pyrrole ring and globin proteins), and iron. Newly formed red blood cells have to get from the bone marrow into a blood vessel. To do this, they squeeze through the endothelial cells of the vessel, but their nucleus cannot fit, so it gets pinched off. The new RBC in the bloodstream has a little bit of endoplasmic reticulum and bits of DNA deposits left over from where the nucleus was pinched off, so a brand new (immature) RBC in the bloodstream is called a reticulocyte. Thus, RBC’s are in an immature state when they are released into the bloodstream. It takes about 1-2 days for these endoplasmic bits to dissolve. Until then, you can see the difference between a reticulocyte and a mature RBC under the microscope when looking at a blood smear. Reticulocytes are immature red blood cells. Only about 1% of the RBC’s should be reticulocytes. If there are more, it indicates a problem. As long as a RBC is flexible, it can weave around the web of reticular fibers in the liver and spleen without getting stuck. Those that get stuck are phagocytized by macrophages. RBC’s that are old or hemoglobin abnormalities (sickle cells) tend to be less flexible, and are caught in reticular fibers and destroyed. This can also happen to reticulocytes. This process takes 7 days: hemocytoblast myeloid stem cell reticulocyte RBC Up until the reticulocyte is released, it retains its shredded endoplasmic reticulum, trying to undergo protein synthesis to make hemoglobin as much as possible. Erythropoiesis is stimulated by a hormone called EPO, which is secreted by the kidney. In conditions where there are not enough RBC’s in the body (e.g. Erythroblastosis Fetalis), oxygen levels will decrease. The kidneys will sense that, and produce EPO, which stimulates stem cells to divide faster, and hastens the release of immature RBCs into the bloodstream, so we will see more reticulocytes in a blood smear. When the RBC is fully mature, it lacks organelles. This is good, because it can transport more O2 if it does not contain any mitochondria, which consume oxygen. However, there is a disadvantage, to not having a nucleus and other organelles: a RBC can’t repair any damage or express new proteins. That’s why they only live 120 days; they accumulate damage, lose their flexibility, and get destroyed. A true RBC count should be at a steady state, new ones replace dying ones. A Hemoglobin molecule consists of three parts 1. Globin chains (proteins from gene expression) 2. Pyrrole ring Heme Group 3. Iron GLOBIN CHAINS To make the globin chains, we need genes. If there is a defect in the gene, the globin chains are defective, as in the case of sickle cell disease. Since it is the iron that binds the oxygen, why do we need globin at all? Because iron binds to oxygen so strongly, it will never let go unless hemoglobin is there to move its structure to block the magnetism of the iron. We need for iron to bind strongly to the oxygen in the lungs. When there is no oxygen on a hemoglobin molecule, the globin chains move a little, exposing the iron so it can grab some oxygen while in the lungs. Once the iron is bound to oxygen, the globin chains move a little, decreasing the hold of iron onto the oxygen, so that the first oxygen-depleted cell that it comes close to can pull the oxygen molecule off of the hemoglobin complex. This is considered reversible binding of oxygen; hemoglobin has an affinity for oxygen in lungs, and low affinity to oxygen in the tissues. There are different types of globin chains. In an adult, there are 2 alpha globin chains and 2 beta chains. Therefore, adult hemoglobin is called A2B2. Since each globin chain is a protein, and there are four proteins bound to each other, hemoglobin has quaternary structure. An embryo has embryonic hemoglobin, called A2E2. An embryo does not have working blood vessels yet, since oxygen is coming in from the placenta. Therefore, an embryo needs Hgb with a higher affinity for oxygen than mom’s A2B2, to rip the oxygen off mom’s hemoglobin. When the embryo develops into a fetus, its blood vessels get bigger, have closer proximity to mom’s blood vessels, needs a little less affinity than embryonic hemoglobin, but the fetus still needs to have hemoglobin that has a higher affinity for oxygen than A2B2, so fetal hemoglobin is A2G2. Around the time of birth, the baby’s hemoglobin becomes A2B2. Once a baby is breathing on its own, it needs hemoglobin with lower affinity. Therefore, the order of affinity for oxygen of the different types of hemoglobin is HgbE (A2E2), HgbF (A2G2), then HgbA (A2B2). HEME GROUP Within each globin chain is a heme group. A heme group consists of a pyrrole ring, with an iron atom in the middle. Since there are four globin chains per hemoglobin molecule, each Hgb has 4 irons. MAKING HEMOGLOBIN Hemoglobin is made in the mitochondria of the erythrocyte while it is developing (in the proerythroblast stage). Once iron is added to the pyrrole ring, the entire structure is called heme. When you add the four globin chains to heme, it is now called hemoglobin. When a macrophage phagocytizes a RBC, the hemoglobin is taken apart into its components. The iron and globin are recycled, but the pyrrole group is cannot be reused, so it needs to be eliminated from the body as a waste product. . We get iron from our diet. It is absorbed from the intestine and released into the plasma, where it binds to a plasma protein called apotransferrin (when it is not bound to iron) or transferrin (when it is bound to iron). Since we have said that the plasma protein has bound to the iron, we will now call it transferrin. The transferrin protein takes the iron to cells in the body that need iron, or the iron is stored intracellularly in two different forms. 1. Ferritin is a protein within a cell that has bound onto the iron. This same protein, when unbound, is called apoferritin. 2. Hemosiderin is a complex in cells that binds to iron and does not release it for use very easily; it is very insoluble. Macrophages that phagocytize RBC’s tend to accumulate hemosiderin deposits. Too much hemosiderin in a cell or in tissues is toxic. IRON HAS SEVERAL DIFFERENT STATES Ferrous (reduced +2) form binds indirectly to oxygen. We need it in this state. Ferric (oxidized +3) form cannot bind to oxygen. Hgb with iron in ferric form is called methemoglobin. We have proteins to convert iron from its ferric to the ferrous state. There are some household products and pesticides that change ferrous to ferric, and our body may not enough available energy to convert it back to the ferrous form we can use. ERYTHROPOIETIN (EPO) EPO (erythropoietin) is a hormone; 90% of EPO is made in the kidney, 10% is made in the liver. It stimulates all the stem cells of blood, many of which develop into RBCs. The RBCs are ready to exit the bone marrow and enter the circulation in 5 days, plus another 2 days of maturation within the circulatory system. If you lose kidney function, you can become anemic. EPO is released by the kidney in response to low oxygen levels in the tissues (hypoxia). Chemotherapy for cancer patients targets rapidly dividing cells, especially the hair (causes hair loss), stomach lining (causes nausea), and the bone marrow (causing low RBC count, and fatigue). They are given artificial EPO to offset the anemia. Athletes might take EPO (illegally) for “blood doping”. It causes an increase in RBC production, leads to more O2, more ATP, more energy, but it thickens blood, can cause heart attack. Sports authorities have been using a drug test on athletes who use a form of EPO made from bacteria; the bacterial particles are detectable on blood tests. Now, these unscrupulous athletes are getting clever: someone has manufactured human EPO that cannot be detected in these drug tests. This human EPO is approved for medicinal use in Europe, and it is in America on the Black Market. So now, sports authorities do a RBC count. If the RBCs are present in excess of set limit before the race starts, they are disqualified. Some of these athletes get away with it by training with human EPO and donating blood before the race. If you want to climb a high mountain, you can’t just climb to the top in one day. There is less oxygen pressure at high altitudes, so RBCs can’t bind oxygen as well. What you do is go to a base camp, part way up the mountain, and stay there for 2 weeks, so the kidney can release EPO to stimulate RBC production. Then you go up the mountain to the next base camp for 2 weeks. Then you can go to the top, when the new cells can bind to oxygen better. Heme group modification and Excretion (Important slide) Three different cells participate in destruction of RBCs RBC Macrophage Hepatocyte When a RBC is old, it gets trapped in the reticular fibers of the spleen or liver, where a macrophage detects it and engulfs it. Within the macrophage, the globin chains, porphyrrin ring, and iron are detached from each other and liberated. What happens to each of these segments? The Iron is released into plasma, apoferrin binds to it (so now the apoferrin is called transferring), it is taken into cells that can use or store it. The iron is stored as ferritin or Hemosiderin. The globin chains (proteins) are hydrolyzed into amino acids (the building blocks of proteins), which are used for synthesis of any other proteins wherever they are needed. The porphyrin ring is converted in the macrophage to pre-bilirubin (uncongugated). It is released into blood, and since it is hydrophobic, it needs albumin as a protein carrier. It is taken to the liver, and enters a hepatocyte (liver cell). Within the hepatocyte, it is conjugated it with glucuronic acid, which makes it hydrophilic. It is then released into the bile duct, enters the intestines. The bilirubin undergoes further conversion before it becomes part of the feces, and is responsible for the brown color. If there is a blockage of the bile duct, it can only exit the body by the urine; it undergoes a different type of conversion, and the urine will be a deep orange-yellow color. Without the brown color in the feces, they will look white. White stools indicate obstruction in the bile duct. Know what parts of a RBC can be recycled: Not all of the heme, but the globin chains, and the iron. There are several different problems that occur when bilirubin is not excreted: all types lead to jaundice. A newborn baby has to make new RBC’s in the liver, and is not able to deal with hemolysis. Hepatocytes are not mature enough to add the glucouronic acid to the porphyrin ring to conjugate it. Adults who have a gall bladder obstruction, causes conjugated bilirubin to be reabsorbed; they need the kidney to deal with it, and the urine becomes orange. How conjugated bilirubin is broken down and excreted When conjugated bilirubin leaves the bile duct and enters the intestines, bacteria in the colon convert it to another type of bilirubin: urobilinogen, which is further metabolized to stercobilinogen, and finally oxidized to stercobilin. This stercobilin gives feces its brown color. If there is unconjugated bilirubin that arrives in the intestine (from an obstruction in the bile duct), it will be absorbed back into the bloodstream (causing jaundice), and the rest is filtered by kidney, converted to another type of bilirubin (urobilin), which causes the urine to turn orange. BRUISES What happens when a bruise goes from purple to green to yellow? When a blood vessel breaks, RBC’s leak out, and macrophages phagocytize them. The macrophage breaks down the hemoglobin in the blood cells into the heme portion, and the globin portion. The globin portion (made of proteins) is broken down into amino acids, which are transported to wherever in the body they are needed to make new proteins. The heme portion is broken down into iron (which is sent to storage or transported to where it is needed) and the pyrrole ring is released into the tissues. There, it is converted to biliverdin, a green type of bilirubin. The biliverdin is then reduced to unconjugated bilirubin (yellow). The unconjugated bilirubin then travels to the liver through the bloodstream. Because this bilirubin is not soluble, however, it is transported through the blood bound to serum albumin. Once it arrives at the liver, it is conjugated with glucuronic acid (to form conjugated bilirubin) to become more water soluble. This conjugated bilirubin is excreted from the liver into the gallbladder and becomes part of bile. Intestinal bacteria convert the bilirubin into urobilinogen. From here the urobilinogen can take two pathways. It can either be further converted into stercobilinogen, which is then oxidized to stercobilin and passed out in the feces, or it can be reabsorbed by the intestinal cells, transported in the blood to the kidneys, and passed out in the urine as the oxidized product urobilin. Stercobilin and urobilin are the products responsible for the color of feces and urine, respectively. Know what happens during hemoglobin breakdown: what happens to all its parts, where does bilirubin go? JAUNDICE When a person has jaundice, is it high levels of conjugated or unconjugated bilirubin? It depends on what is causing the jaundice. Pre-hepatic jaundice is caused by anything which causes an increased rate of hemolysis (breakdown of red blood cells). This can be caused by such things as malaria, sickle cell anemia, Hereditary Spherocytosis, Hemolytic Disease of the Newborn (HDN), glucose 6-phosphate dehydrogenase deficiency (G6DH). Pre-hepatic jaundice will have increased unconjugated bilirubin in the serum. Post-hepatic jaundice, also called obstructive jaundice, is caused by a blockage in the bile duct (portal hypertension), usually by gallstones. Post-hepatic jaundice will have increased conjugated bilirubin in the serum. Hepatic jaundice is from the inability of hepatocytes to conjugate and excrete bilirubin. This includes acute hepatitis, hepatotoxicity and alcoholic liver disease. Hepatic jaundice will have increased unconjugated bilirubin in the serum. In alcoholics, their conjugated bilirubin can also be high. In alcoholics, their unconjugated bilirubin levels are high in the serum because their hepatocytes are damaged. Their conjugated bilirubin levels are high because they also lack albumin, ascites occurs, and the abdominal fluid puts pressure on bile duct, so the conjugated bilirubin is not removed from body. It gets reabsorbed by intestines, and the serum levels of conjugated bilirubin are also increased. Treatment for ascites is to drain it out and take a diuretic. Gotta get the melon tapped! RBC PATHOLOGY 1. Polycythemia (too many RBC’s) 2. Anemia (too few RBC’s) a. Defective i. Iron Deficiency Anemia ii. Aplastic Anemia (cells are not generated) iii. Megaloblastic Anemia iv. Thalassemia v. Hereditary Spherocytosis vi. Sickle Cell Anemia vii. G6PD Deficiency (Glucose 6 Phosphate Deficiency) b. Blood Loss (Hemorrhagic Anemia) i. Traumatic hemorrhage ii. Hemolytic anemia (crosses over into the defective category) 1. Extrinsic (Acquired) 2. Intrinsic (Inherited) POLYCYTHEMIA This condition is too many RBC’s, affects viscosity and blood flow, and causes an increased work load for the heart. The heart needs a more forceful contraction, becomes enlarged over time. It is something to be concerned about, since there is an underlying problem that keeps the RBC’s developing too quickly. The heart can only enlarge safely to a certain limit; after that, it cannot pump properly, and the person can get heart failure. Types of polycythemia: 1. Relative Polycythemia This is a decrease in the plasma volume (dehydration), but the RBC count is normal. The hematocrit will be high, and EPO is normal, since it is the ratio of RBC’s/plasma. Before thinking it is primary polycythemia, check the heart rate, urine output, and ask about dehydration. A dehydrated person will have high hematocrit, low blood pressure, high heart rate, and low urine output. 2. Absolute Polycythemia This is the overproduction of red blood cells, and may be due to a primary process in the bone marrow (a myeloproliferative syndrome), or it may be a reaction to chronically low oxygen levels. a. Primary Polycythemia (Polycythemia Vera, or idiopathic polycythemia) i. Problem with myeloproliferation: There is a problem with stem cells replicating too much (can also affect WBCs). If the problem is in the hemocytoblasts, all cells increased. If just the myelogenous stem cell is a problem , the lymphocytes are not elevated. The hematocrit is high and EPO levels will be low because the kidneys are satisfied with enough oxygen, but the stem cells don’t obey the negative feedback. Kidneys are normal. Treatment is to donate blood frequently, and replace with IV of isosmotic solution. b. Secondary polycythemia i. Problem is a reaction to chronically low oxygen levels, such as during pregnancy, or living in high altitudes. In this case, hematocrit is high and EPO level is high. The kidneys are normal. Treatment is just to monitor the situation for secondary problems: these people need the extra oxygen. ii. Problem in the kidneys, causing inappropriate increase in EPO. May be caused by kidney disorder, tissue hypoxia (from heart or lung disease, including smoking), hepatic problems, or athletes’ blood doping. Hematocrit is high and EPO is high. Treatment is low flow oxygen. ANEMIA Anemia is any condition that causes a reduced oxygen carrying capacity. It is not defined as a low Hct, since that is only one type of anemia. Two categories of anemia: 1. Defective RBC Production a. Dietary (will get better when diet improves) 1) Iron Deficiency Anemia (abnormal Hgb) 2) Megaloblastic Anemia (abnormal cell size) b. Genetic 1) Aplastic Anemia (cells are not generated) 2) Thalassemia (abnormal Hgb) 3) Hereditary Spherocytosis (abnormal membrane) 4) Sickle Cell Anemia (abnormal Hgb) 5) G6PD (Glucose 6 Phosphate) Deficiency (cannot repair own damage) 2. Blood Loss (Hemorrhagic Anemia) a. Traumatic Hemorrhage b. Hemolytic Anemia (genetic defect in Hgb, but considered in this category) i. Extrinsic (Acquired) ii. Intrinsic (Inherited) REVIEW MCH/MCV = MCHC MCV is the blood volume of the blood cell (small =microcytic, large =megaloblastic) MCH is the amount of Hgb in a RBC (low =hypochromic) MCHC is the concentration of Hgb compared to the entire volume of the cell. If you got 50/100 on an exam, your score is 50%. Let’s say this is normal: MCH = 40 MCV = 80 Then MCHC is 40/80 = 50% If cells are megaloblastic (elevated MCV) and normotonic (MCHC is normal), the cell got bigger, but the amount of Hgb must have increased as the cell enlarged. This is the case with megaloblastic anemia. The MCV might be 80 (elevated), the MCV might be 160 (elevated) but the MCHC is 80/160 = 50% (normal). What happens to the MCHC if cells are microcytic (low MCH) and hypochromic (low MCV)? Ratio may be 20/60, so MCHC is low (33%). This is seen in thalassemia and iron deficiency anemia. In some diseases, the person starts out with normochromic and normocytic cells, but with time, the cell shrinks, but hemoglobin stays the same. This will be a low MCH, normal MCV, low MCHC. Example: 20/80 = 25% MCHC. Be mindful of the size, amount of hemoglobin, and how the MCHC will change. IRON DEFICIENCY ANEMIA Either the person does not eat enough iron, or cannot absorb iron from the intestine, or not enough iron is in storage. This causes the hemoglobin to not be made correctly. Cells become microcytic, hypochromic, and low MCH, MCV, MCHC. Children and pregnant women often get this form of anemia. NOTE: Iron pills are bright red, look like candy; be careful! Children can get them and overdose. Iron deficiency anemia often causes weird cravings for things like dirt and charcoal. They often like to chew ice all the time. Anemia is there because there is not enough iron, can’t carry the oxygen. When the cells are too small, their membranes are not properly flexible, get hung up on reticulocytes, and destroyed. MEGALOBLASTIC ANEMIA Caused by lack of dietary vitamin B12 and folic acid A coenzyme is a non-protein chemical compound that binds to a protein and is required for the protein's biological activity. Coenzymes are called "helper molecules" since they assist in biochemical transformations. They help an enzyme to be more efficient. Coenzymes are not proteins, they don’t have amino acids to provide a binding site, unlike an enzyme (which is a catalyst). Many coenzymes are vitamins, or made from vitamins. The coenzyme binds to a different area on the enzyme and causes the enzyme to shift so that its substrate binding site opens up to allow the substrate to bind. The enzyme’s amino acids have to be in correct order to allow the proper binding site for its substrate. The coenzyme does not change. Vitamin B12 and folic acid are coenzymes. They are important for synthesis of thymine (a DNA nucleotide). Lack of thymine still allows DNA replication, but the cell cannot divide, so it gets bigger. Since the cell can use its RNA, the focus is on protein synthesis. Megaloblastic cells get larger, can’t divide, but protein synthesis continues, so there is an increase in Hgb in the cell. MCH and MCV go up, but proportionally to each other. Can get 50/100 and 100/200. Stem cells stay locked in the growth phase. Folic acid Folic acid is actually another B vitamin (B9). It is found in vegetables (especially green leafy ones), fortified grains, and fruits. The liver has several months’ storage for folic acid. People who are often deficient in folic acid are children and pregnant women, people with poor nutrition, alcoholism, or sprue (iliac disease, loss of microvilli in small intestine, don’t absorb as much). Vitamin B12 Vitamin B12 can only be synthesized by bacteria and is found primarily in meat, eggs and dairy products. Because strict vegetarians (vegans) don’t eat these products, they are often deficient in vitamin B12, unless they take it in pill form. Vitamin B12 cannot be absorbed from the intestines to the bloodstream without intrinsic factor, which is secreted by the parietal cells in the stomach. People who have had a gastrectomy also lose their parietal and chief cells. Remember, parietal cells release intrinsic factor and HCL (P I + HCL … don’t get into a “pickle” on a test). Chief cells in the stomach make pepsinogin, and HCl converts that to pepsin (digestive enzyme). Intrinsic factor is needed to allow the small intestine can absorb vitamin B12. People who lack of intrinsic factor from gastric surgery will need weekly vitamin B12 shots for life. They can’t take it orally because it cannot be absorbed without the intrinsic factor. A person who has megaloblastic anemia from a lack of intrinsic factor is said to have a particular type of megaloblastic anemia, called pernicious anemia. The liver has several years’ storage of vitamin B12 (3-5 years). APLASTIC ANEMIA (CELLS NOT GENERATED) 1. Primary aplastic anemia is idiopathic (unknown cause). Stem cells are not replicating enough. Need stem cell transplant. 2. Secondary aplastic anemia can be caused by several things. a. Drugs like antibiotics (chloramphenicol) might cause loss of stem cell production b. Chemicals (pesticides with benzenes) c. Radiation d. A virus e. Malignancy f. Immune suppression g. Decreased EPO from any of the following: Problem with kidney. Rena disease, AIDS, chronic infections, hypometabolic state with protein deprivation, or hypopituitarism. THALASSEMIA 1. Alpha Thalassemia is a mutation in one or more of the gene that makes the alpha globin chains of hemoglobin. You need two alpha globin chains in each hemoglobin molecule. If there are not enough, the Hgb molecule substitutes another beta globin chain, so the hemoglobin is now A1B3 or just A0B4, both of which will precipitate out of solution, interfering with DNA replication, gene expression, and development. Cells from alpha thalassemia are microcytic, hypochromic. Low MCV low MCH, but not proportionally. There are three genes that code for alpha globin, so alpha thalassemia is not common. You get two of these genes from one parent, and two from the other parent. If you have a mutation in one gene, there are three other genes that can make the alpha globin, so there are no problems. But if there are mutations in two of the genes, might have mild episodes of anemia, never diagnosed as thalassemia. If you have 3 genes damaged, will have chronic problem. All 4 genes damaged, will die in utero. 2. Beta Thalassemia has only two genes that code for beta globin. You get one gene from one parent, one from the other parent. A mutation in just one gene can be a problem, so beta thalassemia is more common than alpha thalassemia. Beta thalassemia interferes with cell function. Cells are microcytic, hypochromic, and MCV, MCH, and MCHC are all low. Because these defective cells are phagocytized in the liver and spleen, person gets hepatosplenomegaly. Because these cells are removed from circulation, EPO levels are high, more RBC’s are made, and this causes expansion of intermedulary cavity (where Erythropoiesis is going on), and this causes skeletal abnormalities; also get iron overload and cellular toxicity. In severe cases, there will be reversal to fetal hemoglobin: the gene for gamma globulin chains is turned on again, and the fetal hemoglobin will take the place of the missing alpha chains. Fetal hemoglobin has less affinity for oxygen, but it’s better than nothing. a. Thalassemia Major (Cooley’s anemia): both genes are mutated (homozygous) b. Thalassemia Minor: one gene is mutated (heterozygous). HEREDITARY SPHEROCYTOSIS (HS) HS is the most common inherited blood membrane disorder. Initially, all three values of MCV, MCH, and MCHC are normal. People with HS lack a certain protein, so the membrane becomes smaller, all inside becomes smaller, and since membrane is also an abnormal shape, the cells are phagocytized. MCV decreases, MCH stays the same, MCHC decreases. If the room collapses with the same 100 people in it, everything inside is more concentrated. Know just the end result. SICKLE CELL ANEMIA Sickle Cell Anemia is from a gene mutation that causes a defect in the beta globin chain, not the same mutation as thalassemia. Glutamic acid (polar, hydrophilic amino acid) is replaced by Valine (nonpolar amino acid) in position 6. At first, all three values are normal for MCV, MCH, MCHC. There are no problems as long as the RBC does not sickle. It sickles from being near hypoxic tissue; causes the beta globin chains line up in long rods (change in shape). When the cells are in this sickle shape, it blocks blood vessels, impedes blood flow, generates even more hypoxia, and more sickling. Things that cause tissue hypoxia include being at high altitudes (airplane or mountain climbing), scuba diving, sleeping (breathing is more shallow while sleeping), respiratory or other illness, overexertion. These conditions lead to a sickle crisis, which is very painful. The cells are caught in reticular fibers and removed, resulting in a reduced RBC count, and anemia. Sickle cell trait (heterozygous) vs. Disease (homozygous) The defective gene is on chromosome 11; you get one chromosome 11 from mom and one from dad. If both parents are heterozygous and you inherit two normal chromosomes (25% chance), both beta globin chains are normal, you do not have sickle cell disease or sickle cell trait, and you can make A2B2. However, if you live in Africa, you might die from malaria. That’s why there are not a lot of people in Africa that have both normal chromosomes. Sickle Cell Disease (HgbSS) is when you inherit both abnormal chromosomes (25% chance of this happening from two heterozygous parents). This form is deadly; they tend to die from anemia. Sickle Cell Trait (HgbSA) is when you inherit one normal chromosome and one abnormal (50% chance), you can make a mixture of hemoglobin: some will be A2B2 (normal) and some will have normal alpha chains but the beta chain is affected. This is Sickle Cell Trait, and is not as severe as Sickle Cell Disease. The heterozygous form is often seen in Africans, those from a country that has a lot of malaria. Those who have one bad copy can survive malaria. Those who have two good copies die of malaria. Those with two bad copies die of anemia. How do you tell if a person has the trait or the disease? Run their hemoglobin on electrophoresis gel (Western Blot technique). Normal hemoglobin will travel all the way to the end of the plate. Hemoglobin with one mutation will travel half way down the plate. Hemoglobin with two mutations will travel only part way down the plate. Therefore, heterozygous plates will show two bands, (one that traveled a little, and one that traveled halfway). There is no advantage to having Sickle Cell Disease, but there is one advantage for having Sickle Cell Trait: malaria resistance. Those with Sickle Cell Disease can take a drug (hydroxyuria) to switch expression of genes to make HbF. Malaria Resistance The good thing about having Sickle Cell Trait (the heterozygous form of Sickle Cell Anemia)is having malaria resistance. The bad thing is dealing with frequent hypoxic crisis. Malaria (“bad air”) is a disease caused by plasmodium, a protozoan that is transmitted by mosquitoes. Mosquitoes only live on nectar, except females when they are pregnant. When a mosquito bites one infected person, it takes up the plasmodium, and spreads it to the next person it bites. The organism travels to liver and invades RBCs, where it is hidden from the immune system. It can replicate in there and the immune system cannot detect it. However, its metabolism within the RBC creates hypoxia there, and cell with the Sickle Trait will sickle, get hung up on reticular fibers, and be phagocytized, killing the parasite. Therefore, a person with Sickle Cell Trait has a higher rate of surviving malaria. G6PD DEFICIENCY: GLUCOSE 6 PHOSPHATE DEFICIENCY This is the most inherited enzyme defect, x-linked. RBC’s don’t have organelles, so they are more vulnerable to damage from oxidants, which cause methemoglobin and denaturation of Hgb. Oxidative damage can also cause proteins to cross link with each other, making the RBC membrane less fluid and flexible; they precipitates out of solution and get phagocytized. These cells also can’t carry oxygen because iron has to be in the ferrous 2+ form. Oxidative damage causes ferrous iron to lose an electron, becoming ferric +3, which cannot transport oxygen. Glutathione is an enzyme that helps remove the oxidative damage. Glutathione will donate an electron to reduce ferric iron (+3) to ferrous (+2). The enzyme is present in other cells as well, serving as an antioxidant. It is the oxidative damage repair man. To keep it working (it gives electrons), it needs a supply of electrons. You have to keep it in a reduced state. To do that, it needs glucose 6 phosphate. If you lack that, RBC does not have the enzyme to repair oxidative damage, gets misshaped. People who have g6DP have to watch what they eat. Oxidative drugs (like antimalarials) and foods high in oxidants (such as fava beans) can cause serious health problems. G6PD deficiency is prevalent in the Middle East, where fava beans are commonly part of the diet; those with the enzyme deficiency have to be careful not to partake of that food. BLOOD LOSS: There is a new blood product called plastic blood. It is a water soluble paste, no refrigeration needed! HEMORRHAGIC ANEMIA 1. Acute hemorrhage can lead to hypovolemia and shock. Normocytic, normochromic, normal MCV, MCH, MCHC are normal 2. Chronic hemorrhage (bleeding ulcer from oversecretion of HCL by parietal cells) can lead to microcytic, hypochromic; low MCV, MCH, and possibly low MCHC. RBC’s eventually have to be made smaller and with less hemoglobin because the stomach ulcer diminishes iron absorption. If iron is not absorbed fast enough, they get iron deficiency anemia. Considered hemorrhagic anemia, but can becomes iron deficiency anemia over time. HEMOLYTIC ANEMIA RBC’s rupture inside vessels. 1. Extrinsic (Acquired): not a problem with RBC, something like snake venom is causing the problem, or immune reaction, blood transfusion not matching, or drug reaction. Normal in all three values, MCH, MCH, MCHC. 2. Intrinsic (inherited): problem is how the RBC is made.