
Homologous and Nonhomologous Rearrangements: Interactions
... (positive when realizing a function, negative when inhibiting it). This means that the protein contributes to the range [m−w, m+w] of metabolic processes, with a preference for the processes closest to m (for which the highest efficiency, h, is reached). Thus, various types of proteins can co-exist, ...
... (positive when realizing a function, negative when inhibiting it). This means that the protein contributes to the range [m−w, m+w] of metabolic processes, with a preference for the processes closest to m (for which the highest efficiency, h, is reached). Thus, various types of proteins can co-exist, ...

3 -2 -1 -2 -1 1 2 K
... Definition of Orthology and Paralogy A myoglobin example BLAST search steps Step 1: Specifying Sequence of interest Step 2: Selecting BLAST Program Step 3: Selecting a Database Step 4: Selecting Search Parameters and Formatting Parameters BLAST algorithm uses local alignment search strategy BLAST al ...
... Definition of Orthology and Paralogy A myoglobin example BLAST search steps Step 1: Specifying Sequence of interest Step 2: Selecting BLAST Program Step 3: Selecting a Database Step 4: Selecting Search Parameters and Formatting Parameters BLAST algorithm uses local alignment search strategy BLAST al ...

The Non-LTR Retrotransposon Rex3 from the Fish Xiphophorus is
... Fishes make up more than half of the 48,000 species of living vertebrates. They should therefore possess genetic tools for speciation-associated genome evolution. Transposons may be one of the factors fulfilling this function due to their ability to move within genomes, to generate mutations, and to ...
... Fishes make up more than half of the 48,000 species of living vertebrates. They should therefore possess genetic tools for speciation-associated genome evolution. Transposons may be one of the factors fulfilling this function due to their ability to move within genomes, to generate mutations, and to ...

Slide 1
... Defining a genomic radius for long-range enhancer action: duplicated conserved non-coding elements hold the key. Trends Genet. 2006, 22(1):5-10. ...
... Defining a genomic radius for long-range enhancer action: duplicated conserved non-coding elements hold the key. Trends Genet. 2006, 22(1):5-10. ...

An rpoB signature sequence provides unique resolution for the
... The use of morphological characters for the classification of cyanobacteria has often led to ambiguous strain assignment. In the past two decades, the availability of sequences, such as those of the 16S rRNA, nif, cpc and rpoC1 genes, and the use of metagenomics, has steadily increased and has made ...
... The use of morphological characters for the classification of cyanobacteria has often led to ambiguous strain assignment. In the past two decades, the availability of sequences, such as those of the 16S rRNA, nif, cpc and rpoC1 genes, and the use of metagenomics, has steadily increased and has made ...

Exploring Important Biological Concepts Using Biology Workbench
... Genetic Code: Missense mutations may result in a chemically similar amino acid being inserted at that position in an amino acid chain. Mutations that result in the substitution of one amino acid for another are termed missense mutations. Due to the relationship among codons in the genetic code, many ...
... Genetic Code: Missense mutations may result in a chemically similar amino acid being inserted at that position in an amino acid chain. Mutations that result in the substitution of one amino acid for another are termed missense mutations. Due to the relationship among codons in the genetic code, many ...

Definitions for annotating CDS sequences
... that flank the relevant CDS that need to be analyzed at the nucleotide level, it is sufficient to indicate “N/A”. It is also worth noting that any sequences outside of the linker sequences will be masked out and not analyzed. 5’ Linker – any sequences upstream of the relevant CDS for which the user ...
... that flank the relevant CDS that need to be analyzed at the nucleotide level, it is sufficient to indicate “N/A”. It is also worth noting that any sequences outside of the linker sequences will be masked out and not analyzed. 5’ Linker – any sequences upstream of the relevant CDS for which the user ...

pplacer: linear time maximum-likelihood and Bayesian phylogenetic
... to large volumes of short reads from next-generation sequencing due to computational complexity issues and lack of phylogenetic signal. “Phylogenetic placement,” where a reference tree is fixed and the unknown query sequences are placed onto the tree via a reference alignment, is a way to bring the ...
... to large volumes of short reads from next-generation sequencing due to computational complexity issues and lack of phylogenetic signal. “Phylogenetic placement,” where a reference tree is fixed and the unknown query sequences are placed onto the tree via a reference alignment, is a way to bring the ...

Molecular evidence for the existence of additional members of the
... convalescent phases) and from four healthy controls working in the same workplace were collected. Mononuclear cells purified from 2.5 ml blood using Histopaque (Sigma-Aldrich) were centrifuged in a microcentrifuge and the pellet was processed as described above. Vessel wall tissue was obtained from ...
... convalescent phases) and from four healthy controls working in the same workplace were collected. Mononuclear cells purified from 2.5 ml blood using Histopaque (Sigma-Aldrich) were centrifuged in a microcentrifuge and the pellet was processed as described above. Vessel wall tissue was obtained from ...

Woods Hole – Zebrafish Genetics and Development Bioinformatics
... from analyzing conserved syntenies, in which gene content on particular chromosomes has been retained after species divergence. A useful viewer of conserved syntenies in multiple organisms can be found at the Oxgrid website: http://oxgrid.angis.org.au/oxg_table.html ...
... from analyzing conserved syntenies, in which gene content on particular chromosomes has been retained after species divergence. A useful viewer of conserved syntenies in multiple organisms can be found at the Oxgrid website: http://oxgrid.angis.org.au/oxg_table.html ...

Dell`Orphano: SNP discovery
... • Filter 2: Identifies sequence mismatches by either base substitution type or insertion/deletion type. • Filter 3 & 4: Addressed quality of each base call relative to its position and frequency in a contig. First 100 bases discarded. Consed view of a contig containing a high quality mismatch (A vs. ...
... • Filter 2: Identifies sequence mismatches by either base substitution type or insertion/deletion type. • Filter 3 & 4: Addressed quality of each base call relative to its position and frequency in a contig. First 100 bases discarded. Consed view of a contig containing a high quality mismatch (A vs. ...

The sequence of a gene encoding convicilin from pea
... pea seeds, in addition to legumin and vicilin [1]. It can be purified from both legumin and vicilin, and it consists solely of polypeptides of Mr approx. 71 000. It does not thus contain polypeptides found in either of the two major storage proteins [2]. On the other hand, convicilin is antigenicall ...
... pea seeds, in addition to legumin and vicilin [1]. It can be purified from both legumin and vicilin, and it consists solely of polypeptides of Mr approx. 71 000. It does not thus contain polypeptides found in either of the two major storage proteins [2]. On the other hand, convicilin is antigenicall ...
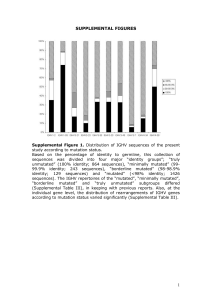
Supplementary Figures (doc 928K)
... examples with real data, namely the formation of clusters 2-0020 and 10089 with four samples each. Black nodes are either clusters (smaller nodes) or samples (larger nodes). Blue lines connect clusters to samples and clusters to clusters, while red lines connect samples between themselves. A. Cluste ...
... examples with real data, namely the formation of clusters 2-0020 and 10089 with four samples each. Black nodes are either clusters (smaller nodes) or samples (larger nodes). Blue lines connect clusters to samples and clusters to clusters, while red lines connect samples between themselves. A. Cluste ...

Why Do More Divergent Sequences Produce Smaller
... properties of the maximum-likelihood estimates of v and d in pairwise alignments and explore the possibility that the negative correlation can be entirely explained by those properties. We show that the v estimate is positively biased for small d and that the bias decreases with the increase of d. W ...
... properties of the maximum-likelihood estimates of v and d in pairwise alignments and explore the possibility that the negative correlation can be entirely explained by those properties. We show that the v estimate is positively biased for small d and that the bias decreases with the increase of d. W ...

2.4. Sequence databases
... be structured based on hierarchical relationships. By doing so, Biological databases programming tasks can be simplified for data that are known to have complex relationships. However, the object-oriented database lacks the rigorous mathematical foundation of the relational databases e.g. it is not ...
... be structured based on hierarchical relationships. By doing so, Biological databases programming tasks can be simplified for data that are known to have complex relationships. However, the object-oriented database lacks the rigorous mathematical foundation of the relational databases e.g. it is not ...

MyTaxa: an advanced taxonomic classifier for genomic and
... or sequence (e.g. the 16S rRNA gene provides robust resolution at the genus level and higher but poor resolution at the species level) are advantageous. However, most, if not all, of the dynamic approaches developed for these purposes rely on some unrealistic assumptions such as that genes of the sa ...
... or sequence (e.g. the 16S rRNA gene provides robust resolution at the genus level and higher but poor resolution at the species level) are advantageous. However, most, if not all, of the dynamic approaches developed for these purposes rely on some unrealistic assumptions such as that genes of the sa ...

Expressed Sequence Tag (EST)
... EST clustering consists in incorporating overlapping ESTs which tag the same Transcript of the same gene in a single cluster For clustering, we measure the similarity (distance) between any 2 sequences. The distance is then reduced to a simple binary value: - accept or reject two sequences in the sa ...
... EST clustering consists in incorporating overlapping ESTs which tag the same Transcript of the same gene in a single cluster For clustering, we measure the similarity (distance) between any 2 sequences. The distance is then reduced to a simple binary value: - accept or reject two sequences in the sa ...

RNA sequencing - Bioinformatics.ca
... • Interpreting mutations that do not have an obvious effect on protein sequence – ‘Regulatory’ mutations that affect what mRNA isoform is expressed and how much • e.g. splice sites, promoters, exonic/intronic splicing motifs, etc. ...
... • Interpreting mutations that do not have an obvious effect on protein sequence – ‘Regulatory’ mutations that affect what mRNA isoform is expressed and how much • e.g. splice sites, promoters, exonic/intronic splicing motifs, etc. ...

Analysis of Similarities/Dissimilarities of DNA Sequences Based on a
... developments of sequencing techniques, many scientists from all kinds of researching fields are attracted to exploit the secrets of life. However, it is difficult to obtain biological informat- ...
... developments of sequencing techniques, many scientists from all kinds of researching fields are attracted to exploit the secrets of life. However, it is difficult to obtain biological informat- ...

Problems 06
... *What microbes will almost certainly be present but unobservable using this approach?* ...
... *What microbes will almost certainly be present but unobservable using this approach?* ...

Problems 07: Metagenomics
... *What microbes will almost certainly be present but unobservable using this approach?* ...
... *What microbes will almost certainly be present but unobservable using this approach?* ...

Innovative Database Models and Advanced Tools in Bioinformatics
... • We have developed and tested a program that considers an ordered set of motifs (or sequence attributes) and searches based on a context of ...
... • We have developed and tested a program that considers an ordered set of motifs (or sequence attributes) and searches based on a context of ...

Impact of Tandem Repeats on the Scaling of Nucleotide Sequences
... ABSTRACT Techniques such as detrended fluctuation analysis (DFA) and its extensions have been widely used to determine the nature of scaling in nucleotide sequences. In this brief communication we show that tandem repeats which are ubiquitous in nucleotide sequences can prevent reliable estimation o ...
... ABSTRACT Techniques such as detrended fluctuation analysis (DFA) and its extensions have been widely used to determine the nature of scaling in nucleotide sequences. In this brief communication we show that tandem repeats which are ubiquitous in nucleotide sequences can prevent reliable estimation o ...

History and Philosophy of Science
... Knowledge and Search in BLAST BLAST differs from many of the informatics tools that we have considered in the course. Essentially it finds a sequence’s nearest neighbors within a database with minimal concern for the content. Unlike discovery or analysis tools, BLAST gathers information and leaves ...
... Knowledge and Search in BLAST BLAST differs from many of the informatics tools that we have considered in the course. Essentially it finds a sequence’s nearest neighbors within a database with minimal concern for the content. Unlike discovery or analysis tools, BLAST gathers information and leaves ...

Recurrence time statistics: Versatile tools for genomic DNA
... Over the past decades, sequence alignment and database search [18–27] have played a significant role in molecular biology, and extensive research in algorithms has resulted in a few basic software tools such as FASTA [28, 29] and BLAST [30–33]. Although these tools have been routinely used in many d ...
... Over the past decades, sequence alignment and database search [18–27] have played a significant role in molecular biology, and extensive research in algorithms has resulted in a few basic software tools such as FASTA [28, 29] and BLAST [30–33]. Although these tools have been routinely used in many d ...
Sequence alignment

In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrix. Gaps are inserted between the residues so that identical or similar characters are aligned in successive columns.Sequence alignments are also used for non-biological sequences, such as those present in natural language or in financial data.