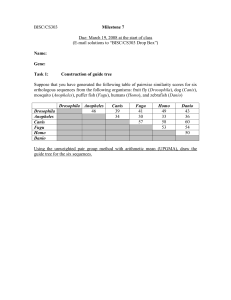
Milestone7
... At the top of the results page, click on the “Start Jalview” button to open an interactive display of the MSA. One of the advantages of a MSA is that it can provide insight into various properties of a family of proteins. When studying your MSA, if you find portions of your sequences that do not ali ...
... At the top of the results page, click on the “Start Jalview” button to open an interactive display of the MSA. One of the advantages of a MSA is that it can provide insight into various properties of a family of proteins. When studying your MSA, if you find portions of your sequences that do not ali ...

Clustered alignments of gene-expression time series data
... Method – SCOW (Shorting COW) • COW (Nielsen et al., 1998) – a dynamic programming algorithm designed to find an optimal alignment between two series with multiple channels of information(such as genes). – Briefly, it aligns and scores two give time series based on their similarity – Two series as q ...
... Method – SCOW (Shorting COW) • COW (Nielsen et al., 1998) – a dynamic programming algorithm designed to find an optimal alignment between two series with multiple channels of information(such as genes). – Briefly, it aligns and scores two give time series based on their similarity – Two series as q ...

Exercise 11 - Understanding the Output for a blastn Search
... character denotes identical bases between the query and the subject sequences. An empty space in the matching sequence denotes a mismatched base. The ‘-‘ character in either the query or the subject sequence denotes a gap in the alignment (Figure 8). ...
... character denotes identical bases between the query and the subject sequences. An empty space in the matching sequence denotes a mismatched base. The ‘-‘ character in either the query or the subject sequence denotes a gap in the alignment (Figure 8). ...

MCSIS - Radboud Universiteit
... Entropy and Variability are two (of the) ways to measure variability patterns. Entropy and Variability patterns can say something about the type of function, and thus add detail to correlation studies. ...
... Entropy and Variability are two (of the) ways to measure variability patterns. Entropy and Variability patterns can say something about the type of function, and thus add detail to correlation studies. ...

BioPHP - Minitools Chaos Game Representation of DNAGraphical
... DNA sequence manipulation/properties This program has multiple functions. Using this tool, a variety of routine DNA manipulation tasks can be performed such as, removing the non-coding characters in the sequence, reversing the sequence, reverse complement, to show the complementary strand sequence, ...
... DNA sequence manipulation/properties This program has multiple functions. Using this tool, a variety of routine DNA manipulation tasks can be performed such as, removing the non-coding characters in the sequence, reversing the sequence, reverse complement, to show the complementary strand sequence, ...

Name: : - Ms. Poole`s Biology
... and sequencing the resulting small fragments. The entire protein sequence is then reconstructed by matching regions of sequence overlap seen in the small fragments. Algorithms may be used to compare protein sequence data against a database. These algorithms provide local information (within a defin ...
... and sequencing the resulting small fragments. The entire protein sequence is then reconstructed by matching regions of sequence overlap seen in the small fragments. Algorithms may be used to compare protein sequence data against a database. These algorithms provide local information (within a defin ...

Pair-wise sequence alignment
... For comparing long DNA sequences with a short one Comparing a gene with a complete genome For detecting similarities between highly diverged sequences which still share common subsequences (that have little or no mutations). ...
... For comparing long DNA sequences with a short one Comparing a gene with a complete genome For detecting similarities between highly diverged sequences which still share common subsequences (that have little or no mutations). ...

Slide 1
... ProSite Pattern Search Profile Search Hidden Markov Models (HMMs) Domain (Pfam); Whole protein (PIRSF) Neural Networks ...
... ProSite Pattern Search Profile Search Hidden Markov Models (HMMs) Domain (Pfam); Whole protein (PIRSF) Neural Networks ...

Tutorial 7: Constructing new databases using ARB
... ARB is most frequently utilized for management and analysis of SSU rRNA gene data, but it can be a very useful tool to align, manage, and compare sequence data from other genes. The features used for analysis of SSU rRNA genes are very similar as to working with other genes, but one difference is th ...
... ARB is most frequently utilized for management and analysis of SSU rRNA gene data, but it can be a very useful tool to align, manage, and compare sequence data from other genes. The features used for analysis of SSU rRNA genes are very similar as to working with other genes, but one difference is th ...

09_01.jpg
... MSA Approaches • Progressive approach CLUSTALW (CLUSTALX) PILEUP T-COFFEE • Iterative approach: Repeatedly realign subsets of sequences. MultAlin, DiAlign. ...
... MSA Approaches • Progressive approach CLUSTALW (CLUSTALX) PILEUP T-COFFEE • Iterative approach: Repeatedly realign subsets of sequences. MultAlin, DiAlign. ...

Lecture 27
... • Realistic way of assigning the probability of occurrence (weight) for a substitution is to look at the physical similarity of amino acids. • Dayhoff measured a number of residue exchanges for closely related proteins and determined their relative frequency of the 20 X 19/2 = 190 different possible ...
... • Realistic way of assigning the probability of occurrence (weight) for a substitution is to look at the physical similarity of amino acids. • Dayhoff measured a number of residue exchanges for closely related proteins and determined their relative frequency of the 20 X 19/2 = 190 different possible ...

- BioMed Central
... A copy of the scripts used by ROSLIN The following script takes a list of accession numbers and uses then to retrieve fasta sequence files for each gene using the emboss software package. The sequences are then blasted against the latest version of the pig genome (7) which was downloaded from the Sa ...
... A copy of the scripts used by ROSLIN The following script takes a list of accession numbers and uses then to retrieve fasta sequence files for each gene using the emboss software package. The sequences are then blasted against the latest version of the pig genome (7) which was downloaded from the Sa ...


Particle Mesh Ewald(PME) method
... to work on the same task By dividing the database into roughly equal chunks, we naturally solve any load imbalance problems This technique works for uniform recurrence equations Ubiquitous in computational biology including local sequence alignment, multiple sequence alignment, motif finding ...
... to work on the same task By dividing the database into roughly equal chunks, we naturally solve any load imbalance problems This technique works for uniform recurrence equations Ubiquitous in computational biology including local sequence alignment, multiple sequence alignment, motif finding ...

Exercises
... Άσκηση 4 Ανάλυση ακολουθιών πυρηνικών οξέων χρησιμοποιώντας το Internet Sequence analysis using Internet functions Short tutorial on restriction mapping, translation, and BLAST. Many of the following exercises involve copying one sequence from a page in Netscape to another. For these types of exerci ...
... Άσκηση 4 Ανάλυση ακολουθιών πυρηνικών οξέων χρησιμοποιώντας το Internet Sequence analysis using Internet functions Short tutorial on restriction mapping, translation, and BLAST. Many of the following exercises involve copying one sequence from a page in Netscape to another. For these types of exerci ...

Molecular Phylogenetic Analysis: Design and Implementation of
... have been developed and published to solve the different problems involved in these studies. Once the dataset of biological sequences has been selected, the first step is to align them [1,2]. The difference in lengths can appear due to sequencing errors (digitalizing the biological sample), mutation ...
... have been developed and published to solve the different problems involved in these studies. Once the dataset of biological sequences has been selected, the first step is to align them [1,2]. The difference in lengths can appear due to sequencing errors (digitalizing the biological sample), mutation ...

ppt
... sequences that are greater than >6% are from different species • Using models based on a poisson distribution and 3 different coverage models, estimates of species for the whole study range from 1800 to 47,000. • A minimum of 12X greater sequence effort would be needed to sample 95% of the unique se ...
... sequences that are greater than >6% are from different species • Using models based on a poisson distribution and 3 different coverage models, estimates of species for the whole study range from 1800 to 47,000. • A minimum of 12X greater sequence effort would be needed to sample 95% of the unique se ...


lecture05_11
... – ref: CLUSTAL: a package for performing multiple sequence alignment on a microcomputer. Gene, 73, 237–244. [Medline] ...
... – ref: CLUSTAL: a package for performing multiple sequence alignment on a microcomputer. Gene, 73, 237–244. [Medline] ...

Visualization of Biological Sequence Similarity Search
... shift [18]. Biologists are interested in frame shifts, especially since they are very important to the function of the sequence and are difficult to discover. In this example, the first green/yellow region seemsto encode one protein in one frame, and the second red region seems to encode a different ...
... shift [18]. Biologists are interested in frame shifts, especially since they are very important to the function of the sequence and are difficult to discover. In this example, the first green/yellow region seemsto encode one protein in one frame, and the second red region seems to encode a different ...

Challenge Lesson Analyzing DNA
... 2. In the search window at the top of the page, first select the word “Nucleotide” from the pull-down bar. Then, in the text box next to the pull-down bar, type in “Monodelphis domestica low density lipoprotein receptor.” Click on the first result that appears: “AY871266.1”. (Alternatively, you can ...
... 2. In the search window at the top of the page, first select the word “Nucleotide” from the pull-down bar. Then, in the text box next to the pull-down bar, type in “Monodelphis domestica low density lipoprotein receptor.” Click on the first result that appears: “AY871266.1”. (Alternatively, you can ...

Validity of transferring the footprint sites identified in lab
... A concern was raised that laboratory lines might be subject to conditions that may have led to the loss of functional binding sites, either by selection or by genetic drift. If so, the footprint site data on lab liines might not be representative of natural population sequences. The effect of a non- ...
... A concern was raised that laboratory lines might be subject to conditions that may have led to the loss of functional binding sites, either by selection or by genetic drift. If so, the footprint site data on lab liines might not be representative of natural population sequences. The effect of a non- ...
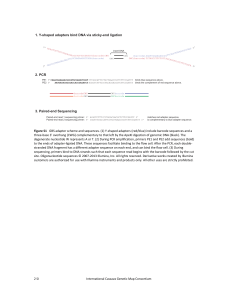
Figure S1 - G3: Genes | Genomes | Genetics
... degenerate nucleotide W represents A or T. (2) During PCR amplification, primers PE1 and PE2 add sequences (bold) to the ends of adapter‐ligated DNA. These sequences facilitate binding to the flow cell. After the PCR, each double‐ stranded DNA fragment has a different adapter sequence on each end, ...
... degenerate nucleotide W represents A or T. (2) During PCR amplification, primers PE1 and PE2 add sequences (bold) to the ends of adapter‐ligated DNA. These sequences facilitate binding to the flow cell. After the PCR, each double‐ stranded DNA fragment has a different adapter sequence on each end, ...

... • The first biological database - Protein Identification Resource was established in 1972 by Margaret Dayhoff • Dayhoff and co-workers organized the proteins into families and superfamilies based on degree of sequence similarity • Idea of sequence alignment was introduced as well as special tables t ...

Poster
... this end, a score is attributed to each locus in the genome according to the similarity measure defined by the matrix. The output of this functionality is filtered with a cut-off score and then directly used as input by the second one. The second functionality starts by fetching the gene positions o ...
... this end, a score is attributed to each locus in the genome according to the similarity measure defined by the matrix. The output of this functionality is filtered with a cut-off score and then directly used as input by the second one. The second functionality starts by fetching the gene positions o ...
Sequence alignment

In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrix. Gaps are inserted between the residues so that identical or similar characters are aligned in successive columns.Sequence alignments are also used for non-biological sequences, such as those present in natural language or in financial data.