
Bio 125 Lab Week 9: Relating Changes in Form to Changes in Genes
... in Column 3 of the data table. (You should have filled out the second column of Table 4 already, as described on page 1.) You’ll find this information handy when you look at your tree. Note that all eight sequence names end with “cauliflower,” which refers to the name of the gene, not the subspecies ...
... in Column 3 of the data table. (You should have filled out the second column of Table 4 already, as described on page 1.) You’ll find this information handy when you look at your tree. Note that all eight sequence names end with “cauliflower,” which refers to the name of the gene, not the subspecies ...

CSE527 Project Report
... tend to occur frequently outside promoter regions, resulting in a large number of false positives. Another reason is that usually multiple transcription factors act in concert. Their motifs have to be aligned within a certain distance and often ordering. These cis-regulatory modules (CRM’s) are typi ...
... tend to occur frequently outside promoter regions, resulting in a large number of false positives. Another reason is that usually multiple transcription factors act in concert. Their motifs have to be aligned within a certain distance and often ordering. These cis-regulatory modules (CRM’s) are typi ...

Expressed Sequence Tag (EST)
... EST clustering consists in incorporating overlapping ESTs which tag the same Transcript of the same gene in a single cluster For clustering, we measure the similarity (distance) between any 2 sequences. The distance is then reduced to a simple binary value: - accept or reject two sequences in the sa ...
... EST clustering consists in incorporating overlapping ESTs which tag the same Transcript of the same gene in a single cluster For clustering, we measure the similarity (distance) between any 2 sequences. The distance is then reduced to a simple binary value: - accept or reject two sequences in the sa ...

Evolution of Disintegrin Cysteine-Rich and
... the Clustal V program (Higgins et al. 1992). Progressive alignments used the multiple alignment algorithms described by Higgins and Sharp (1989), with a fixed gap penalty of 10 and the Dayhoff PAM 250 protein weight matrix (Dayhoff et al. 1978). Alignments were finally refined by eye and differ very ...
... the Clustal V program (Higgins et al. 1992). Progressive alignments used the multiple alignment algorithms described by Higgins and Sharp (1989), with a fixed gap penalty of 10 and the Dayhoff PAM 250 protein weight matrix (Dayhoff et al. 1978). Alignments were finally refined by eye and differ very ...

Compressed q-gram Indexing for Highly Repetitive Biological
... Pair compression [12], and use it to search for the desired q-grams. The index is able to search for pattern strings of any length, and particularly effective on short ones. Our experimental results show that both techniques are competitive with the state of the art in this particular scenario, whe ...
... Pair compression [12], and use it to search for the desired q-grams. The index is able to search for pattern strings of any length, and particularly effective on short ones. Our experimental results show that both techniques are competitive with the state of the art in this particular scenario, whe ...

Sequencing project for Bi1x
... Alignment of 16S sequences is different than alignment of protein coding nucleotide sequences because the 16S aligners take into account the secondary structure of the 16S gene. For example, sequences on both sides of a stem loop need to match up in order to form the stem loop. Greengenes also attem ...
... Alignment of 16S sequences is different than alignment of protein coding nucleotide sequences because the 16S aligners take into account the secondary structure of the 16S gene. For example, sequences on both sides of a stem loop need to match up in order to form the stem loop. Greengenes also attem ...

Presentation
... Multialignments • Select BLAST alignments to be multialigned • Clustal-W performs multialignment • Aligns – The originating IMAS gene sequence – The “Full” sequence found by BLAST • Not just the high-quality section – Useful to align entire genes, or entire corresponding segments of DNA ...
... Multialignments • Select BLAST alignments to be multialigned • Clustal-W performs multialignment • Aligns – The originating IMAS gene sequence – The “Full” sequence found by BLAST • Not just the high-quality section – Useful to align entire genes, or entire corresponding segments of DNA ...

Slide 1
... (BLAST) finds regions of local similarity between sequences. The program compares nucleotide or protein sequences to sequence databases and calculates the statistical significance of matches. BLAST can be used to infer functional and evolutionary relationships between sequences as well as help ident ...
... (BLAST) finds regions of local similarity between sequences. The program compares nucleotide or protein sequences to sequence databases and calculates the statistical significance of matches. BLAST can be used to infer functional and evolutionary relationships between sequences as well as help ident ...
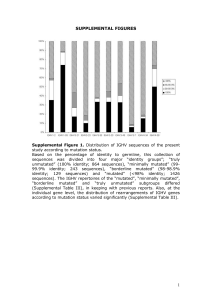
Supplementary Figures (doc 928K)
... Supplemental Figure 5. A Biolayout 2D representation of two clustering examples with real data, namely the formation of clusters 2-0020 and 10089 with four samples each. Black nodes are either clusters (smaller nodes) or samples (larger nodes). Blue lines connect clusters to samples and clusters to ...
... Supplemental Figure 5. A Biolayout 2D representation of two clustering examples with real data, namely the formation of clusters 2-0020 and 10089 with four samples each. Black nodes are either clusters (smaller nodes) or samples (larger nodes). Blue lines connect clusters to samples and clusters to ...

Slide 1
... Defining a genomic radius for long-range enhancer action: duplicated conserved non-coding elements hold the key. Trends Genet. 2006, 22(1):5-10. ...
... Defining a genomic radius for long-range enhancer action: duplicated conserved non-coding elements hold the key. Trends Genet. 2006, 22(1):5-10. ...

pdf
... ultra-conserved elements is highly improbable in neutrally evolving regions. 22.1 The data Our analyses of ultra-conserved elements are based on multiple sequence alignments produced by MAVID [Bray and Pachter, 2004]. Prior to the alignment of multiple genomes, homology mappings (from Mercator [Dewe ...
... ultra-conserved elements is highly improbable in neutrally evolving regions. 22.1 The data Our analyses of ultra-conserved elements are based on multiple sequence alignments produced by MAVID [Bray and Pachter, 2004]. Prior to the alignment of multiple genomes, homology mappings (from Mercator [Dewe ...

Dell`Orphano: SNP discovery
... • Filter 2: Identifies sequence mismatches by either base substitution type or insertion/deletion type. • Filter 3 & 4: Addressed quality of each base call relative to its position and frequency in a contig. First 100 bases discarded. Consed view of a contig containing a high quality mismatch (A vs. ...
... • Filter 2: Identifies sequence mismatches by either base substitution type or insertion/deletion type. • Filter 3 & 4: Addressed quality of each base call relative to its position and frequency in a contig. First 100 bases discarded. Consed view of a contig containing a high quality mismatch (A vs. ...

Why Do More Divergent Sequences Produce Smaller
... (v = dN/dS) and the sequence distance d in pairwise comparisons of the same gene from different species. That is, more divergent sequences produce smaller estimates of v. Explanations for this negative correlation have included segregating nonsynonymous polymorphisms in closely related species and n ...
... (v = dN/dS) and the sequence distance d in pairwise comparisons of the same gene from different species. That is, more divergent sequences produce smaller estimates of v. Explanations for this negative correlation have included segregating nonsynonymous polymorphisms in closely related species and n ...

Discovery of Cyclotide-Like Protein Sequences in Graminaceous
... The cyclotides are the largest known family of circular proteins (Craik et al., 1999). They contain ;30 amino acids, including six conserved Cys residues that are linked in pairs to form three disulfide bonds. The three disulfide bonds are connected in a knotted topology in which one disulfide threa ...
... The cyclotides are the largest known family of circular proteins (Craik et al., 1999). They contain ;30 amino acids, including six conserved Cys residues that are linked in pairs to form three disulfide bonds. The three disulfide bonds are connected in a knotted topology in which one disulfide threa ...

Surveying Saccharomyces Genomes to Identify Functional Elements
... likely encode small proteins (<100 amino acids) that have not been annotated. Comparisons of S. cerevisiae intergenic sequences to those of the other Saccharomyces species using TBLASTX revealed many potential protein-coding sequences. Such comparisons of the sensu stricto sequences are not very inf ...
... likely encode small proteins (<100 amino acids) that have not been annotated. Comparisons of S. cerevisiae intergenic sequences to those of the other Saccharomyces species using TBLASTX revealed many potential protein-coding sequences. Such comparisons of the sensu stricto sequences are not very inf ...

pplacer: linear time maximum-likelihood and Bayesian phylogenetic
... means of understanding the evolutionary origin of query sequences. The presence of a query sequence on a certain branch of a tree gives precise information about the evolutionary relationship of that sequence to other sequences in the tree. For example, a query sequence placed deep in the tree can i ...
... means of understanding the evolutionary origin of query sequences. The presence of a query sequence on a certain branch of a tree gives precise information about the evolutionary relationship of that sequence to other sequences in the tree. For example, a query sequence placed deep in the tree can i ...

Milestone3
... sequence of a motif, we would miss other (degenerate) instances of the motif. How then might we search for an instance of a gene’s TATA box, if the instance might differ from the consensus sequence? One approach would be to search for sequences of six nucleotides either that match the consensus sequ ...
... sequence of a motif, we would miss other (degenerate) instances of the motif. How then might we search for an instance of a gene’s TATA box, if the instance might differ from the consensus sequence? One approach would be to search for sequences of six nucleotides either that match the consensus sequ ...

DNA Sequence Capture and Enrichment by Microarray Followed by
... potential for the efficient enrichment of specific, large high-complexity genomic regions of interest (14, 15, 19, 20 ). Combining this technology with NGS produces a powerful sequencing tool that has the potential to be implemented in a clinical diagnostics laboratory. Aside from such issues as tes ...
... potential for the efficient enrichment of specific, large high-complexity genomic regions of interest (14, 15, 19, 20 ). Combining this technology with NGS produces a powerful sequencing tool that has the potential to be implemented in a clinical diagnostics laboratory. Aside from such issues as tes ...

Database resources of the National Center for Biotechnology
... selected for bulk download. A BLAST variant, BLAST2Sequences (11), compares two DNA or protein sequences and produces a dot-plot representation of the alignments. Each alignment returned by a BLAST search receives a score and a measure of statistical signi®cance, called the Expectation Value (E-valu ...
... selected for bulk download. A BLAST variant, BLAST2Sequences (11), compares two DNA or protein sequences and produces a dot-plot representation of the alignments. Each alignment returned by a BLAST search receives a score and a measure of statistical signi®cance, called the Expectation Value (E-valu ...

Considerations for Analyzing Targeted NGS Data – HLA
... Lots of similar genes and lots of very similar pseudegenes. Duplicated segments can be more similar to each other within an individual than they are similar to the corresponding segments of the reference genome. ...
... Lots of similar genes and lots of very similar pseudegenes. Duplicated segments can be more similar to each other within an individual than they are similar to the corresponding segments of the reference genome. ...

Spider Silk - Consortium for Mathematics and its Applications
... means, proteins. They continue to develop new and better techniques. For example, given a protein sequence that we would like to synthesize, it is possible to “program” microorganisms to synthesize these proteins for us. Scientists do this by building a DNA sequence that codes for the desired protei ...
... means, proteins. They continue to develop new and better techniques. For example, given a protein sequence that we would like to synthesize, it is possible to “program” microorganisms to synthesize these proteins for us. Scientists do this by building a DNA sequence that codes for the desired protei ...

Course Form - Bluegrass Community and Technical College
... Determine protein coding regions and putative protein sequence(s) within a DNA sequence using existing bioinformatic databases. Perform searches of nucleotide and protein databases using a query sequence and retrieve sequences that are related to the query sequence. Align and compare multiple DNA or ...
... Determine protein coding regions and putative protein sequence(s) within a DNA sequence using existing bioinformatic databases. Perform searches of nucleotide and protein databases using a query sequence and retrieve sequences that are related to the query sequence. Align and compare multiple DNA or ...

Why do more divergent sequences produce smaller non
... Several studies have reported a negative correlation between estimates of the nonsynonymous to synonymous rate ratio (ω = dN /dS ) and the sequence distance d in pairwise comparisons of the same gene from different species. That is, more divergent sequences produce smaller estimates of ω. Explanatio ...
... Several studies have reported a negative correlation between estimates of the nonsynonymous to synonymous rate ratio (ω = dN /dS ) and the sequence distance d in pairwise comparisons of the same gene from different species. That is, more divergent sequences produce smaller estimates of ω. Explanatio ...


Types of variation in DNA-A among isolates of East African cassava
... genes apparently can have a different evolutionary origin from complementary-sense genes, and ori seems to be a recombination hot spot, occurs among begomoviruses infecting cotton and okra in Pakistan (Zhou et al., 1998). If the same is true for EACMV, the extent of the sequence dissimilarity betwee ...
... genes apparently can have a different evolutionary origin from complementary-sense genes, and ori seems to be a recombination hot spot, occurs among begomoviruses infecting cotton and okra in Pakistan (Zhou et al., 1998). If the same is true for EACMV, the extent of the sequence dissimilarity betwee ...
Multiple sequence alignment

A multiple sequence alignment (MSA) is a sequence alignment of three or more biological sequences, generally protein, DNA, or RNA. In many cases, the input set of query sequences are assumed to have an evolutionary relationship by which they share a lineage and are descended from a common ancestor. From the resulting MSA, sequence homology can be inferred and phylogenetic analysis can be conducted to assess the sequences' shared evolutionary origins. Visual depictions of the alignment as in the image at right illustrate mutation events such as point mutations (single amino acid or nucleotide changes) that appear as differing characters in a single alignment column, and insertion or deletion mutations (indels or gaps) that appear as hyphens in one or more of the sequences in the alignment. Multiple sequence alignment is often used to assess sequence conservation of protein domains, tertiary and secondary structures, and even individual amino acids or nucleotides.Multiple sequence alignment also refers to the process of aligning such a sequence set. Because three or more sequences of biologically relevant length can be difficult and are almost always time-consuming to align by hand, computational algorithms are used to produce and analyze the alignments. MSAs require more sophisticated methodologies than pairwise alignment because they are more computationally complex. Most multiple sequence alignment programs use heuristic methods rather than global optimization because identifying the optimal alignment between more than a few sequences of moderate length is prohibitively computationally expensive.