
Department of Health Information Management
... Pairwise Local Alignment • Pairwise local sequence alignment: identify similar segments in two sequences • Smith-Waterman algorithm (a dynamic programming algorithm) is guaranteed to find optimal alignments, but it is computationally expensive [O(nm)]. ...
... Pairwise Local Alignment • Pairwise local sequence alignment: identify similar segments in two sequences • Smith-Waterman algorithm (a dynamic programming algorithm) is guaranteed to find optimal alignments, but it is computationally expensive [O(nm)]. ...

final_report_columns
... substitutions and predicts whether an amino acid substitution in a protein will have a phenotypic effect. SIFT is based on the premise that protein evolution is correlated with protein function. Positions important for function should be conserved in an alignment of the protein family, whereas unimp ...
... substitutions and predicts whether an amino acid substitution in a protein will have a phenotypic effect. SIFT is based on the premise that protein evolution is correlated with protein function. Positions important for function should be conserved in an alignment of the protein family, whereas unimp ...

Bioinformatics Resources at a Glance A Note about FASTA Format
... A Note about FASTA Format There are MANY free bioinformatics tools available online. Bioinformaticists have developed a standard format for nucleotide and protein sequences that allows them to be read by a wide range of programs. This format is called FASTA format, and each nucleotide or amino ac ...
... A Note about FASTA Format There are MANY free bioinformatics tools available online. Bioinformaticists have developed a standard format for nucleotide and protein sequences that allows them to be read by a wide range of programs. This format is called FASTA format, and each nucleotide or amino ac ...

Gentile, Margaret: Computational Methods for the Design of PCR Primers for the Amplification of functional Markers from Environmental Samples
... Challenges of primer design for unknown, diverse sequences The design of a primer to amplify a gene of interest from all species present differs from the applications described above, because the sequence to be amplified is not actually known and can be quite different from known sequences of the ge ...
... Challenges of primer design for unknown, diverse sequences The design of a primer to amplify a gene of interest from all species present differs from the applications described above, because the sequence to be amplified is not actually known and can be quite different from known sequences of the ge ...


Supplementary Information
... 3. Quantitative RT-PCR. Patient muscle RNA was prepared from ~100 mg of vastus lateralis biopsy material by homogenising in 1 ml of QIAzol (RNA extraction kit, QIAGEN) and subjecting to RNA extraction according to the manufacturers’ instructions. Human muscle RNA purchased from Ambion was used as a ...
... 3. Quantitative RT-PCR. Patient muscle RNA was prepared from ~100 mg of vastus lateralis biopsy material by homogenising in 1 ml of QIAzol (RNA extraction kit, QIAGEN) and subjecting to RNA extraction according to the manufacturers’ instructions. Human muscle RNA purchased from Ambion was used as a ...

lab6
... • MEME (Multiple EM for Motif Elicitation) discovers patterns in nucleotide and amino acid sequences. • MEME paper • Few tutorials - 1( Pg 6 ), 2, 3, 4 ...
... • MEME (Multiple EM for Motif Elicitation) discovers patterns in nucleotide and amino acid sequences. • MEME paper • Few tutorials - 1( Pg 6 ), 2, 3, 4 ...


Comparative genomics exercises - Genome curation on emerging
... * The sequences are “Locked” i.e. they will scroll together. At the bottom left corner you will see the word ‘LOCKED’. To allow the sequences to scroll independently of each other, they need to be “unlocked”. To unlock, right-click anywhere on the screen and uncheck “Lock Sequence”. # If red connect ...
... * The sequences are “Locked” i.e. they will scroll together. At the bottom left corner you will see the word ‘LOCKED’. To allow the sequences to scroll independently of each other, they need to be “unlocked”. To unlock, right-click anywhere on the screen and uncheck “Lock Sequence”. # If red connect ...

doc
... The workbook is organised around 5 practical exercises that use databases and software to solve problems associated with GFP sequence data: 1: Use of databases, (a-c) to confirm the identity of clones and search for similar nucleotide and translated protein sequences and alignment of sequences 2: Id ...
... The workbook is organised around 5 practical exercises that use databases and software to solve problems associated with GFP sequence data: 1: Use of databases, (a-c) to confirm the identity of clones and search for similar nucleotide and translated protein sequences and alignment of sequences 2: Id ...

Similarity Searches on Sequence Databases: BLAST
... • However this assumption doesn’t hold all the time, some sequences have biased compositions, e.g. many proteins contain patches known as low-complexity regions: such as segments that contain many prolines or glutamic acid residues. • If BLAST aligns two proline-rich domains, this alignment gets a v ...
... • However this assumption doesn’t hold all the time, some sequences have biased compositions, e.g. many proteins contain patches known as low-complexity regions: such as segments that contain many prolines or glutamic acid residues. • If BLAST aligns two proline-rich domains, this alignment gets a v ...

XLibraryDisplay User Manual Ryan Stafford
... were run to determine background levels for the assay. It is ok to have multiple identical sample IDs with asterisks since they do not need to be uniquely associated with sequences. The program will check your data for consistency or other issues when you try to correlate sequences to activity data, ...
... were run to determine background levels for the assay. It is ok to have multiple identical sample IDs with asterisks since they do not need to be uniquely associated with sequences. The program will check your data for consistency or other issues when you try to correlate sequences to activity data, ...

Methods for pattern discovery in unaligned biological sequences
... to build a model for the source generating the pattern. Then, for each input sequence, the substring that best ®ts the model is considered the occurrence of the corresponding pattern. Further details on how models are represented and evaluated will be shown in the section on `Measures of signi®cance ...
... to build a model for the source generating the pattern. Then, for each input sequence, the substring that best ®ts the model is considered the occurrence of the corresponding pattern. Further details on how models are represented and evaluated will be shown in the section on `Measures of signi®cance ...

Annotation report - GEP Community Server
... 3. Alignment between the submitted model and the D. melanogaster ortholog Show an alignment between the protein sequence for your gene model and the protein sequence from the putative D. melanogaster ortholog. You can either use the protein alignment generated by the Gene Model Checker (available th ...
... 3. Alignment between the submitted model and the D. melanogaster ortholog Show an alignment between the protein sequence for your gene model and the protein sequence from the putative D. melanogaster ortholog. You can either use the protein alignment generated by the Gene Model Checker (available th ...

Bioinformatics Supplement - Bio-Rad
... perform the same function as the protein produced by the daf-18 gene? We can answer these questions by delving into data stored in large genetics databases like those maintained by the National Center for Biotechnology Information (NCBI), such as GenBank. By performing a homology search — a search i ...
... perform the same function as the protein produced by the daf-18 gene? We can answer these questions by delving into data stored in large genetics databases like those maintained by the National Center for Biotechnology Information (NCBI), such as GenBank. By performing a homology search — a search i ...
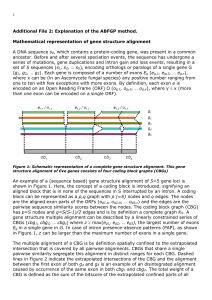
Abstract - BioMed Central
... At this stage all true CBGs can be correctly retrieved. This is only valid for genecontaining sequences that have a high level of protein similarity over their complete lengths, that lack small exons and lack small CBGs. Regions of little similarity, occurrence of small exons (5~20 nt) and intron-ex ...
... At this stage all true CBGs can be correctly retrieved. This is only valid for genecontaining sequences that have a high level of protein similarity over their complete lengths, that lack small exons and lack small CBGs. Regions of little similarity, occurrence of small exons (5~20 nt) and intron-ex ...

Name that Gene Project The National Center for Biotechnology
... EXERCISE 1: From the main BLAST page select Nucleotide BLAST. This brings up a web page where you can specify your query sequence along with various parameters. Copy and paste the above "dinosaur DNA" sequence into the window labeled Enter Query Sequence, and then click the BLAST button at the botto ...
... EXERCISE 1: From the main BLAST page select Nucleotide BLAST. This brings up a web page where you can specify your query sequence along with various parameters. Copy and paste the above "dinosaur DNA" sequence into the window labeled Enter Query Sequence, and then click the BLAST button at the botto ...

introduction to molecular phylogeny
... the alignment (site) is supposed to contain homologous residues (nucleotides, amino acids) that derive from a common ancestor. ==> Unreliable parts of the alignment must be omitted from further phylogenetic analysis. ...
... the alignment (site) is supposed to contain homologous residues (nucleotides, amino acids) that derive from a common ancestor. ==> Unreliable parts of the alignment must be omitted from further phylogenetic analysis. ...

Nucleotide substitutions and evolution of duplicate genes.
... In a large-scale analysis involving several pairs of genes the sequences in one pair have a different length than sequences in another pair. Thus the goal is to compute the number of substitutions per site or number of substitutions per codon. In our search for duplicate genes we used two different ...
... In a large-scale analysis involving several pairs of genes the sequences in one pair have a different length than sequences in another pair. Thus the goal is to compute the number of substitutions per site or number of substitutions per codon. In our search for duplicate genes we used two different ...

Comparison of Genomes using High-Performance - FACOM
... genomes not handled by the previous work. More specifically, we locate and compare not only the homologous genes (expressed in terms of the 20-letter amino acids) but also compare the regions or gaps (in terms of the 4-letter DNA nucleotides) between the corresponding homologous ...
... genomes not handled by the previous work. More specifically, we locate and compare not only the homologous genes (expressed in terms of the 20-letter amino acids) but also compare the regions or gaps (in terms of the 4-letter DNA nucleotides) between the corresponding homologous ...

Introduction to Biological Data
... Why Databases? • The purpose of databases is not merely to collect and organize data, but to allow intelligent data retrieval. • A query is a method to retrieve information from the database. • The organization of each record into predetermined fields, allows us to use queries on fields. ...
... Why Databases? • The purpose of databases is not merely to collect and organize data, but to allow intelligent data retrieval. • A query is a method to retrieve information from the database. • The organization of each record into predetermined fields, allows us to use queries on fields. ...

BLAST Exercise: Detecting and Interpreting Genetic Homology
... The Basic Local Alignment Search Tool (BLAST) is a program that reports regions of local similarity (at either the nucleotide or protein level) between a query sequence and sequences within a database. The ability to detect sequence homology allows us to determine if a gene or a protein is related t ...
... The Basic Local Alignment Search Tool (BLAST) is a program that reports regions of local similarity (at either the nucleotide or protein level) between a query sequence and sequences within a database. The ability to detect sequence homology allows us to determine if a gene or a protein is related t ...

Unoshan_project
... directions, and the bases of the individual nucleotides are on the inside of the helix, stacked on top of each other. The larger base shape shown in the picture above depicts the purines (adenine and guanine). The other base shape in the illustration represents the pyrimidines (cytosine and thymine) ...
... directions, and the bases of the individual nucleotides are on the inside of the helix, stacked on top of each other. The larger base shape shown in the picture above depicts the purines (adenine and guanine). The other base shape in the illustration represents the pyrimidines (cytosine and thymine) ...

Gene Hunting for the Cystic Fibrosis gene - CusMiBio
... colored segments, the name and the access number of the corresponding sequence will appear; clicking on one of the segments will take you to the corresponding sequence alignment; • under the graphic scheme you find a new link: “distance tree of result” which is a graphic representation of the evolut ...
... colored segments, the name and the access number of the corresponding sequence will appear; clicking on one of the segments will take you to the corresponding sequence alignment; • under the graphic scheme you find a new link: “distance tree of result” which is a graphic representation of the evolut ...

Construction of PANM Database (Protostome DB) for rapid
... molluscan sequence information especially the EST sequences generated by traditional sequencing methods. However, it is found that the server has limitations in the annotation of molluscan sequences generated using next-generation sequencing (NGS) platforms due to inconsistencies in molluscan sequen ...
... molluscan sequence information especially the EST sequences generated by traditional sequencing methods. However, it is found that the server has limitations in the annotation of molluscan sequences generated using next-generation sequencing (NGS) platforms due to inconsistencies in molluscan sequen ...
Multiple sequence alignment

A multiple sequence alignment (MSA) is a sequence alignment of three or more biological sequences, generally protein, DNA, or RNA. In many cases, the input set of query sequences are assumed to have an evolutionary relationship by which they share a lineage and are descended from a common ancestor. From the resulting MSA, sequence homology can be inferred and phylogenetic analysis can be conducted to assess the sequences' shared evolutionary origins. Visual depictions of the alignment as in the image at right illustrate mutation events such as point mutations (single amino acid or nucleotide changes) that appear as differing characters in a single alignment column, and insertion or deletion mutations (indels or gaps) that appear as hyphens in one or more of the sequences in the alignment. Multiple sequence alignment is often used to assess sequence conservation of protein domains, tertiary and secondary structures, and even individual amino acids or nucleotides.Multiple sequence alignment also refers to the process of aligning such a sequence set. Because three or more sequences of biologically relevant length can be difficult and are almost always time-consuming to align by hand, computational algorithms are used to produce and analyze the alignments. MSAs require more sophisticated methodologies than pairwise alignment because they are more computationally complex. Most multiple sequence alignment programs use heuristic methods rather than global optimization because identifying the optimal alignment between more than a few sequences of moderate length is prohibitively computationally expensive.