
Lecture 2
... What is a genetic network? – Individual genes have a function (e.g. transforming a substance or binding to a substance) – Sets of functions when sequenced can produce pathways (e.g. output of one transformation is the input to another) – Sets of pathways, as they interact with other pathways, create ...
... What is a genetic network? – Individual genes have a function (e.g. transforming a substance or binding to a substance) – Sets of functions when sequenced can produce pathways (e.g. output of one transformation is the input to another) – Sets of pathways, as they interact with other pathways, create ...

Promoter Analysis for Intestinally
... iii. Each result set was extracted independently, so these hits overlap with each other in the original sequence and appear varying numbers of times iv. It is not at all clear which of these hits we should use to form a Position Frequency Matrix and which ones we should discard Future Work 1. Positi ...
... iii. Each result set was extracted independently, so these hits overlap with each other in the original sequence and appear varying numbers of times iv. It is not at all clear which of these hits we should use to form a Position Frequency Matrix and which ones we should discard Future Work 1. Positi ...

MCSIS - Radboud Universiteit
... validated, understood, and used to answer our questions. Now was in 2007… ...
... validated, understood, and used to answer our questions. Now was in 2007… ...

Detection of Transcription Factor Binding Sites
... which can be used by a cell to construct proteins Each set of instructions within this sequence is called a gene ...
... which can be used by a cell to construct proteins Each set of instructions within this sequence is called a gene ...

Bioinformatics Overview, NCBI & GenBank
... • Initially built and maintained at Los Alamos National Laboratory. • Transferred to NCBI in early 1990s by congressional mandate. • Most journal publishers require deposition of sequence data into GanBank prior to publication so an accession number may be cited. • Submitters may keep their data con ...
... • Initially built and maintained at Los Alamos National Laboratory. • Transferred to NCBI in early 1990s by congressional mandate. • Most journal publishers require deposition of sequence data into GanBank prior to publication so an accession number may be cited. • Submitters may keep their data con ...
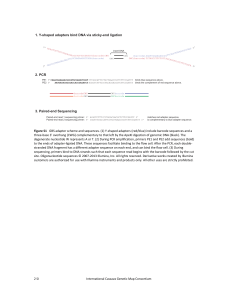
Figure S1 - G3: Genes | Genomes | Genetics
... three‐base 3’ overhang (CWG) complementary to that left by the ApeKI digestion of genomic DNA (black). The degenerate nucleotide W represents A or T. (2) During PCR amplification, primers PE1 and PE2 add sequences (bold) to the ends of adapter‐ligated DNA. These sequences facilitate binding to the ...
... three‐base 3’ overhang (CWG) complementary to that left by the ApeKI digestion of genomic DNA (black). The degenerate nucleotide W represents A or T. (2) During PCR amplification, primers PE1 and PE2 add sequences (bold) to the ends of adapter‐ligated DNA. These sequences facilitate binding to the ...

Align the DNA sequences
... DNA SEQUENCE RESOURCES: The National Center for Biotechnology Information (NCBI)Established in 1988 as a national resource for molecular biology information, NCBI creates public databases, conducts research in computational biology, develops software tools for analyzing genome data, and disseminate ...
... DNA SEQUENCE RESOURCES: The National Center for Biotechnology Information (NCBI)Established in 1988 as a national resource for molecular biology information, NCBI creates public databases, conducts research in computational biology, develops software tools for analyzing genome data, and disseminate ...

Exercises
... Short tutorial on restriction mapping, translation, and BLAST. Many of the following exercises involve copying one sequence from a page in Netscape to another. For these types of exercises, therefore, it is a good idea to use multiple windows of Netscape. To create a new window select File - New Web ...
... Short tutorial on restriction mapping, translation, and BLAST. Many of the following exercises involve copying one sequence from a page in Netscape to another. For these types of exercises, therefore, it is a good idea to use multiple windows of Netscape. To create a new window select File - New Web ...

Molecular Phylogeny
... DNA yields more phylogenetic information than proteins. The nucleotide sequences of a pair of homologous genes have a higher information content than the amino acid sequences of the corresponding proteins, because mutations that result in synonymous changes alter the DNA sequence but do not affect t ...
... DNA yields more phylogenetic information than proteins. The nucleotide sequences of a pair of homologous genes have a higher information content than the amino acid sequences of the corresponding proteins, because mutations that result in synonymous changes alter the DNA sequence but do not affect t ...

ppt
... Regulatory elements evolution Understanding the mechanisms of gene regulation, and how evolution of the pattern of gene regulation contributes to morphological and phenotypic differences among organisms are fundamentally important goals in the genome era ...
... Regulatory elements evolution Understanding the mechanisms of gene regulation, and how evolution of the pattern of gene regulation contributes to morphological and phenotypic differences among organisms are fundamentally important goals in the genome era ...

Comparative Analysis
... BLAST® (Basic Local Alignment Search Tool) is a set of similarity search programs designed to explore all of the available sequence databases regardless of whether the query is protein or DNA. The BLAST programs have been designed for speed, with a minimal sacrifice of sensitivity to distant sequenc ...
... BLAST® (Basic Local Alignment Search Tool) is a set of similarity search programs designed to explore all of the available sequence databases regardless of whether the query is protein or DNA. The BLAST programs have been designed for speed, with a minimal sacrifice of sensitivity to distant sequenc ...

CS790 – Introduction to Bioinformatics
... Global alignments – score the entire alignment Semi-global alignments – allow unscored gaps at the beginning or end of either sequence Local alignment – find the best matching subsequence CGATG AAATGGA ...
... Global alignments – score the entire alignment Semi-global alignments – allow unscored gaps at the beginning or end of either sequence Local alignment – find the best matching subsequence CGATG AAATGGA ...

Particle Mesh Ewald(PME) method
... • Each class is described by short characteristic sequences or motifs • Each class or “generator” is described by a Hidden Markov model • Protein Motif Finding answers the questions: Given a sequence what class does it belong to? Given a sequence and a HMM what is the probability that the sequen ...
... • Each class is described by short characteristic sequences or motifs • Each class or “generator” is described by a Hidden Markov model • Protein Motif Finding answers the questions: Given a sequence what class does it belong to? Given a sequence and a HMM what is the probability that the sequen ...


Sequence
... the top scoring region, it does not locate all high scoring alignments between two sequences. As a consequence, FASTA may not directly identify repeats or multiple domains that are shared between two proteins BLAST - a faster alternative BLAST (Basic Local Alignment Search Tool) is a heuristic metho ...
... the top scoring region, it does not locate all high scoring alignments between two sequences. As a consequence, FASTA may not directly identify repeats or multiple domains that are shared between two proteins BLAST - a faster alternative BLAST (Basic Local Alignment Search Tool) is a heuristic metho ...

CS790 – Introduction to Bioinformatics
... The draft human genome is available Automated gene finding is possible Gene: AGTACGTATCGTATAGCGTAA • What does it do? ...
... The draft human genome is available Automated gene finding is possible Gene: AGTACGTATCGTATAGCGTAA • What does it do? ...

Blast intro slides ppt
... homologous genes and proteins Homologous Proteins (or genes) • Have a common ancestor (they’re related) • Have similar structures • Have similar functions ...
... homologous genes and proteins Homologous Proteins (or genes) • Have a common ancestor (they’re related) • Have similar structures • Have similar functions ...

BLAST intro slides ppt
... homologous genes and proteins Homologous Proteins (or genes) • Have a common ancestor (they’re related) • Have similar structures • Have similar functions ...
... homologous genes and proteins Homologous Proteins (or genes) • Have a common ancestor (they’re related) • Have similar structures • Have similar functions ...

Lecture 27
... Figure 7-28 A guide to the significance of normalized alignment scores (NAS) in the comparison of peptide sequences. ...
... Figure 7-28 A guide to the significance of normalized alignment scores (NAS) in the comparison of peptide sequences. ...

scores
... The Smith-Waterman algorithm is a local alignment tool used to obtain sensitive pairwise similarity alignments. Smith-Waterman algorithm uses dynamic programming. Operating via a matrix, the algorithm uses backtracing and tests alternative paths to the highest scoring alignments, and selects the opt ...
... The Smith-Waterman algorithm is a local alignment tool used to obtain sensitive pairwise similarity alignments. Smith-Waterman algorithm uses dynamic programming. Operating via a matrix, the algorithm uses backtracing and tests alternative paths to the highest scoring alignments, and selects the opt ...

Step 3. Construction of the phylogenetic tree Distance methods
... Therefore, methods have been developed to compensate for this. ...
... Therefore, methods have been developed to compensate for this. ...

class10
... • We wish to assign a probability to each alignment of two DNA/protein sequences, using the HMM model. • Each “output symbol” of the HMM is an aligned pair of two letters, or of a letter and a gap. • The hidden states should represent some evolutionary model. • Transition and emission probabilities ...
... • We wish to assign a probability to each alignment of two DNA/protein sequences, using the HMM model. • Each “output symbol” of the HMM is an aligned pair of two letters, or of a letter and a gap. • The hidden states should represent some evolutionary model. • Transition and emission probabilities ...

Text S1. Predicted Functional RNAs Within Coding Regions
... to produce p-values based on estimated false positive rates [6]. For example, the RNAz manual states that a p-value > 0.5 should result in an approximately 4% false positive rate while a p-value of 0.9 should result in false positive rate of ~ 1%. However, these estimations are based on an artificia ...
... to produce p-values based on estimated false positive rates [6]. For example, the RNAz manual states that a p-value > 0.5 should result in an approximately 4% false positive rate while a p-value of 0.9 should result in false positive rate of ~ 1%. However, these estimations are based on an artificia ...

... • The first biological database - Protein Identification Resource was established in 1972 by Margaret Dayhoff • Dayhoff and co-workers organized the proteins into families and superfamilies based on degree of sequence similarity • Idea of sequence alignment was introduced as well as special tables t ...

Document
... Your MSA should have as few gaps as possible. Some variability but not too much! Some conservation but not too much! ...
... Your MSA should have as few gaps as possible. Some variability but not too much! Some conservation but not too much! ...
Multiple sequence alignment

A multiple sequence alignment (MSA) is a sequence alignment of three or more biological sequences, generally protein, DNA, or RNA. In many cases, the input set of query sequences are assumed to have an evolutionary relationship by which they share a lineage and are descended from a common ancestor. From the resulting MSA, sequence homology can be inferred and phylogenetic analysis can be conducted to assess the sequences' shared evolutionary origins. Visual depictions of the alignment as in the image at right illustrate mutation events such as point mutations (single amino acid or nucleotide changes) that appear as differing characters in a single alignment column, and insertion or deletion mutations (indels or gaps) that appear as hyphens in one or more of the sequences in the alignment. Multiple sequence alignment is often used to assess sequence conservation of protein domains, tertiary and secondary structures, and even individual amino acids or nucleotides.Multiple sequence alignment also refers to the process of aligning such a sequence set. Because three or more sequences of biologically relevant length can be difficult and are almost always time-consuming to align by hand, computational algorithms are used to produce and analyze the alignments. MSAs require more sophisticated methodologies than pairwise alignment because they are more computationally complex. Most multiple sequence alignment programs use heuristic methods rather than global optimization because identifying the optimal alignment between more than a few sequences of moderate length is prohibitively computationally expensive.