
Protein folding
... works of Shakespeare but just the short sentence 'Methinks it is like a weasel', and we shall make it relatively easy by giving him a typewriter with a restricted keyboard, one with just the 26 (capital) letters, and a space bar. How long will he take to ...
... works of Shakespeare but just the short sentence 'Methinks it is like a weasel', and we shall make it relatively easy by giving him a typewriter with a restricted keyboard, one with just the 26 (capital) letters, and a space bar. How long will he take to ...

Definition (956.3 KB)
... are more than one and one-half kilometres long. Both sides of these dunes have practically the same slope and are usually covered with grass and shrubs. While the crest is usually bare of vegetation, on some dunes, even the crest is covered. Where this occurs, it means that wind action has stopped o ...
... are more than one and one-half kilometres long. Both sides of these dunes have practically the same slope and are usually covered with grass and shrubs. While the crest is usually bare of vegetation, on some dunes, even the crest is covered. Where this occurs, it means that wind action has stopped o ...


Protein Structure Analysis and Prediction
... asset. It would help in understanding the structures and functions of the thousands of sequences that are being discovered every day in biotechnology labs [Chan and Dill 1993]. However, predicting tertiary structure from primary structure has proved to be a very difficult problem. This paper describ ...
... asset. It would help in understanding the structures and functions of the thousands of sequences that are being discovered every day in biotechnology labs [Chan and Dill 1993]. However, predicting tertiary structure from primary structure has proved to be a very difficult problem. This paper describ ...

HHMI meeting, FOLDING
... resembles melting or sublimation of a crystal rather than evaporation of a liquid. ...
... resembles melting or sublimation of a crystal rather than evaporation of a liquid. ...

Supplementary Tables and Figures Legends (doc 39K)
... Sepharose beads (Rix, 2007) are shown in blue. The five compounds, being drugs themselves, were chosen for their properties such as size and polarity, but also for their lack of known mammalian targets (e.g. the antibiotic ciprofloxacin) or knowledge that, through coupling to the resin, binding to k ...
... Sepharose beads (Rix, 2007) are shown in blue. The five compounds, being drugs themselves, were chosen for their properties such as size and polarity, but also for their lack of known mammalian targets (e.g. the antibiotic ciprofloxacin) or knowledge that, through coupling to the resin, binding to k ...

Solution structure of the Drosha double-stranded RNA-binding domain Open Access
... upon RNA binding [19]. A model for RNA recognition suggests that the two domains bind to portions of the pri-miRNA that are distant from each other. It is not known whether the dsRBD of Drosha is also important for substrate RNA binding or serves another function, since little to no RNA-binding acti ...
... upon RNA binding [19]. A model for RNA recognition suggests that the two domains bind to portions of the pri-miRNA that are distant from each other. It is not known whether the dsRBD of Drosha is also important for substrate RNA binding or serves another function, since little to no RNA-binding acti ...

PPT
... • short regions of similarity between a pair of sequences. • compared sequences can receive high local similarity scores, without the need to have high levels of similarity over their entire length • useful when looking for domains within proteins or looking for regions of genomic DNA that contain c ...
... • short regions of similarity between a pair of sequences. • compared sequences can receive high local similarity scores, without the need to have high levels of similarity over their entire length • useful when looking for domains within proteins or looking for regions of genomic DNA that contain c ...

Insights From The Molecular Docking Of
... residues. Figure 1B shows the three-dimensional structure (highlighted) of one of the SAAR’s sequences “GGGGGG” observed in the crystal structure (PDB-id: 2X4M) of the plasminogen activator PLA from Yersinia pestis. The amino acid sequence of imidazole nepropionase (Uniprot accession No. Q8U8Z6; len ...
... residues. Figure 1B shows the three-dimensional structure (highlighted) of one of the SAAR’s sequences “GGGGGG” observed in the crystal structure (PDB-id: 2X4M) of the plasminogen activator PLA from Yersinia pestis. The amino acid sequence of imidazole nepropionase (Uniprot accession No. Q8U8Z6; len ...
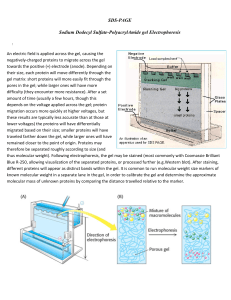
SDS-PAGE Sodium Dodecyl Sulfate
... difficulty (they encounter more resistance). After a set amount of time (usually a few hours, though this depends on the voltage applied across the gel; protein migration occurs more quickly at higher voltages, but these results are typically less accurate than at those at lower voltages) the protei ...
... difficulty (they encounter more resistance). After a set amount of time (usually a few hours, though this depends on the voltage applied across the gel; protein migration occurs more quickly at higher voltages, but these results are typically less accurate than at those at lower voltages) the protei ...

Predicting RNA Secondary Structures
... Hairpins are usually mad of a stem and a loop as shown here ...
... Hairpins are usually mad of a stem and a loop as shown here ...

Lecture 3
... 1. Deformation is forced because of tertiary structure (crowding). 2. Strong H-bonding (e.g., between side chains). 3. Helix breakers inside; Pro will result in a kink for sure and Gly almost always but small polar amino acids such as Ser and Thr also can. ...
... 1. Deformation is forced because of tertiary structure (crowding). 2. Strong H-bonding (e.g., between side chains). 3. Helix breakers inside; Pro will result in a kink for sure and Gly almost always but small polar amino acids such as Ser and Thr also can. ...


seminario Archetti 10-05-17
... so-called RNA Partial Degradation Problem (RNA PDP) which is to reconstruct the original molecule by determining from this limited information the exact positions of the primary and secondary cleavage sites. By solving this combinatorial problem, one can reconstruct a given RNA molecule, having as i ...
... so-called RNA Partial Degradation Problem (RNA PDP) which is to reconstruct the original molecule by determining from this limited information the exact positions of the primary and secondary cleavage sites. By solving this combinatorial problem, one can reconstruct a given RNA molecule, having as i ...

Protein Folding, Shape, and Function Activity Instructions
... A chart of the amino acid sidechains and their properties is available for reference. 5. In addition to using the principles above to fold your protein, also form a pocket or groove (representing an active site) around a small lego (representing the substrate). This active site and substrate should ...
... A chart of the amino acid sidechains and their properties is available for reference. 5. In addition to using the principles above to fold your protein, also form a pocket or groove (representing an active site) around a small lego (representing the substrate). This active site and substrate should ...

Viral Structure Lec. 2
... – Capsid is the storage site for genome – Many capsids have a ‘shell’ structure – Genome + Capsid = Nucleocapsid – Capsid is made up of polymeric proteins to conserve genome • Ex. 5 Kb genome requires 30,000 a/a capsid, which means 90 Kb genome just for capsid!! • Solution: use multiple copies of sa ...
... – Capsid is the storage site for genome – Many capsids have a ‘shell’ structure – Genome + Capsid = Nucleocapsid – Capsid is made up of polymeric proteins to conserve genome • Ex. 5 Kb genome requires 30,000 a/a capsid, which means 90 Kb genome just for capsid!! • Solution: use multiple copies of sa ...

Document
... • Can define most similar regions in a set of proteins – functional domains – structural domains ...
... • Can define most similar regions in a set of proteins – functional domains – structural domains ...

The Art of Multiple Sequence Alignment in R
... of DNA, RNA, or amino acid (AA) sequences and returns a merged alignment. For more than two sequences, the function AlignSeqs can be used to perform multiple sequence alignment in a progressive/iterative manner on sequences of the same kind. In this case, multiple alignment works by aligning two seq ...
... of DNA, RNA, or amino acid (AA) sequences and returns a merged alignment. For more than two sequences, the function AlignSeqs can be used to perform multiple sequence alignment in a progressive/iterative manner on sequences of the same kind. In this case, multiple alignment works by aligning two seq ...
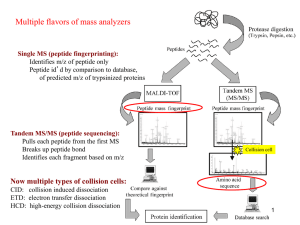
Proteomics2_2012
... - expensive to make many heavy peptides of precise abundance - limited number of proteins that can be analyzed ...
... - expensive to make many heavy peptides of precise abundance - limited number of proteins that can be analyzed ...
Protein Structure Prediction
... Two feed-forward back-propagation networks with a single hidden layer are used where the first sequence-structure network is trained with the multiple sequence alignment in the form of PSI-BLAST generated position specific scoring matrices. ...
... Two feed-forward back-propagation networks with a single hidden layer are used where the first sequence-structure network is trained with the multiple sequence alignment in the form of PSI-BLAST generated position specific scoring matrices. ...

Document
... The covalent bonds to maintain the primary structure peptide bonds, disulfide bonds The peptide chain is known as the backbone, and the "R" groups are known as side chains. The primary structure is usually shown using abbreviations (three letters or one letter) for the amino acid residues,fr ...
... The covalent bonds to maintain the primary structure peptide bonds, disulfide bonds The peptide chain is known as the backbone, and the "R" groups are known as side chains. The primary structure is usually shown using abbreviations (three letters or one letter) for the amino acid residues,fr ...

search1
... • The first round of PSI-BLAST is a standard protein-protein BLAST search. The program builds a position-specific scoring matrix (PSSM or profile) from an alignment of the sequences returned with Expect values better (lower) than the inclusion threshold (default=0.005). • The PSSM will be used to ev ...
... • The first round of PSI-BLAST is a standard protein-protein BLAST search. The program builds a position-specific scoring matrix (PSSM or profile) from an alignment of the sequences returned with Expect values better (lower) than the inclusion threshold (default=0.005). • The PSSM will be used to ev ...

A. Major Histocompatibility Complex (MHC)
... This should open a new menu box with various options for coloring. Select the atoms/bonds radio button and then by element button. This should color the selected atoms by CPK coloring (blue for N, red for O, white for H etc. initially proposed by Robert Corey and Linus Pauling, and improved by Walte ...
... This should open a new menu box with various options for coloring. Select the atoms/bonds radio button and then by element button. This should color the selected atoms by CPK coloring (blue for N, red for O, white for H etc. initially proposed by Robert Corey and Linus Pauling, and improved by Walte ...
Document
... 1) Forms protein’s initial S-S bonds in similar way (protein –SH attacks PDI S-S bond to give mixed disulfide) 2) Protein SH attacks protein-PDI mixed S-S bond to give protein S-S bond 3) Continues until protein in native S-S configuration and PDI cannot bind to exposed hydrophobic patches on the pr ...
... 1) Forms protein’s initial S-S bonds in similar way (protein –SH attacks PDI S-S bond to give mixed disulfide) 2) Protein SH attacks protein-PDI mixed S-S bond to give protein S-S bond 3) Continues until protein in native S-S configuration and PDI cannot bind to exposed hydrophobic patches on the pr ...
Structural alignment

Structural alignment attempts to establish homology between two or more polymer structures based on their shape and three-dimensional conformation. This process is usually applied to protein tertiary structures but can also be used for large RNA molecules. In contrast to simple structural superposition, where at least some equivalent residues of the two structures are known, structural alignment requires no a priori knowledge of equivalent positions. Structural alignment is a valuable tool for the comparison of proteins with low sequence similarity, where evolutionary relationships between proteins cannot be easily detected by standard sequence alignment techniques. Structural alignment can therefore be used to imply evolutionary relationships between proteins that share very little common sequence. However, caution should be used in using the results as evidence for shared evolutionary ancestry because of the possible confounding effects of convergent evolution by which multiple unrelated amino acid sequences converge on a common tertiary structure.Structural alignments can compare two sequences or multiple sequences. Because these alignments rely on information about all the query sequences' three-dimensional conformations, the method can only be used on sequences where these structures are known. These are usually found by X-ray crystallography or NMR spectroscopy. It is possible to perform a structural alignment on structures produced by structure prediction methods. Indeed, evaluating such predictions often requires a structural alignment between the model and the true known structure to assess the model's quality. Structural alignments are especially useful in analyzing data from structural genomics and proteomics efforts, and they can be used as comparison points to evaluate alignments produced by purely sequence-based bioinformatics methods.The outputs of a structural alignment are a superposition of the atomic coordinate sets and a minimal root mean square deviation (RMSD) between the structures. The RMSD of two aligned structures indicates their divergence from one another. Structural alignment can be complicated by the existence of multiple protein domains within one or more of the input structures, because changes in relative orientation of the domains between two structures to be aligned can artificially inflate the RMSD.