Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Discovery and development of proton pump inhibitors wikipedia , lookup
Polysubstance dependence wikipedia , lookup
Orphan drug wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
Psychopharmacology wikipedia , lookup
Compounding wikipedia , lookup
Plateau principle wikipedia , lookup
Theralizumab wikipedia , lookup
Pharmacognosy wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Neuropharmacology wikipedia , lookup
Drug design wikipedia , lookup
Pharmaceutical industry wikipedia , lookup
Prescription costs wikipedia , lookup
Drug discovery wikipedia , lookup
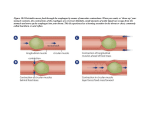
Year Level 6 [Module 2: General Concepts of Disease and Therapeutics] PRINCIPLES OF PHARMACOKINETICS (1) Marcelo Imasa, MD | [Pharmacology] OUTLINE: I. INTRODUCTION A. DEFINITION B. PROCESSES II. LIBERATION A. MODIFIED RELEASE VS. IMMEDIATE RELEASE DRUG PRODUCTS B. POTENCY AND POTENTIATION III. TRANSPORT PROCESSES A. PASSIVE TRANSPORT B. CARRIER-MEDIATED TRANSPORT C. CONVECTIVE TRANSPORT D. PINOCYTOSIS IV. ABSORPTION A. DEFINITIONS B. BIOAVAILABILITY C. BIOEQUIVALENCE D. FACTORS AFFECTING ABSORPTION I. II. July 8, 2009 o o A. May also be non-intentional – poor quality drug products (ex. Cloxacillin – highly hygroscopic that if not properly formulated, it liquefies during storage then eventually solidifies that the drug will no longer be absorbed) If intentional, it is called modified-release product (ER = extended release, SR = sustained release) Modified Release vs. Immediate Release drug products Compared using drug plasma concentration vs time graph Get volunteers to take drugs, then measure the levels in the plasma (y-axis) as function of time (x-axis) Graph 1. Plasma Concentration of Drug vs Time INTRODUCTION Pharmacokinetics – what the body does to the drug Includes the processes the drug undergoes as it reaches the biological site of action and leaves the body LADMER system o Liberation o (Transport) o Absorption o Distribution o Metabolism o Excretion Elimination is the general process to refer to both metabolism and excretion LIBERATION Drugs are usually NOT in pure form Drug product = drug (active) + excipients (substances added to produce a form that can be delivered to the patient) Liberation – is the process of release of the drug from the drug product Objective of liberation: to make the drug available in an aqueous solution form o Why? o Practically all the transport processes require that the substance be in aqueous solution form Exceptions among the drug products taken P.O.: syrups, elixirs – active ingredients are already in aqueous solution (Note: PO means per orem or taken orally and swallowed Liberation is a highly modifiable process o You can change how the drug is released from the drug product o May be intentional - designed by whoever formulated the drug Where: MEC (minimum effective concentration) - the smallest concentration of the drug in the blood that produces a response or effect MTC (minimum toxic concentration) – the upper range of the therapeutic range; the minimum concentration that can produce toxicity to the body Therapeutic Range – area between MTC and MEC Onset – time when concentration reaches MEC Offset – time when concentration goes below MEC Duration of Action – interval between Onset and Offset of drug action; effectivity time Aim of manufacturers: drug concentration in the blood reaches the MEC without reaching the MTC With modified release: o The concentration of the drug between the ONSET and OFFSET reaches a plateau instead of a peak thus there is less likelihood of reaching the MTC compared to immediate release (Example: with immediate release, an anti-diabetic drug can reach MTC easily and cause hypoglycemia. With modified release, the incidence of hypoglycemia is decreased) Team 7 | Agustin, Aranjuez, Magat, Maglaque, Ocampo, Parco, Regalado, Serrano, Tan, Tanbonliong PRINCIPLS OF PHARMACOKINETICS (1) Year Level 6 [Module 2: General Concepts of Disease and Therapeutics] July 8, 2009 o There can be a delay in the onset of action environment of the stomach, the weak acid but longer duration of action drug will undergo higher rates of passive transport assuming all other factors to be Immediate release = usually how drugs are equal discovered (no modifications done) o In an acidic environment, with a weak base It is mandatory that the pattern of release, if it is drug, the reaction will also shift to the left, modified, is put in drug labels towards the ionized form of the base (BH+). o XR = extended release, SR = sustained This lowers the rate of transport of the release, PR = Prolonged-release, etc weakly basic drug in the stomach. B. Potency and Potentiation o Conversely, in a basic environment, the Different from patterns of release non-ionized form of the weakly basic drug Potency - dose that produces 50% of maximum effect (B) is favored, resulting to high transport Potentiation – making the drug “more potent”; done rates of the drug in the small intestines by lowering the dose needed to achieve the same In short, weak acids are optimally absorbed in the maximum effect stomach and weak bases are optimally absorbed in the small intestines (assuming all other factors are III. TRANSPORT PROCESSES equal) B. Carrier-mediated Transport E. Passive Transport A number of drugs in the market requires carriers movement along a concentration gradient; NOT Carriers are proteins with specific binding sites and energy-requiring; SLOW but DOMINANT transport will undergo conformational change when bound process which enables movement of the drug inside the cell Requirements for drugs to undergo passive transport Types o Small (MW<400-600) o Active transport – requires energy, movement is o Lipophilic – lipid-soluble (since the plasma against the electrochemical gradient membrane consists of lipids) Ex. Na+/K+ ATPase Governing principle: Fick’s law of diffusion – rate of o Facilitated diffusion – no energy input, transport is directly proportional to the surface area, movement is down the electrochemical gradient diffusion coefficient and concentration gradient and Ex. GLUT4 – insulin-sensitive glucose transporter inversely proportional to the thickness of the for the permeation of glucose across a muscle membrane cell membrane Common features 1. Selectivity/specificity 2. Subject to competition/ inhibition/ antagonism Where Ex. Isoniazid (anti-TB drug) is associated with a A= surface area side-effect – peripheral neuropathy, which can (C1 – C2)=concentration gradient/difference be resolved by Vitamin B6. They have similarity h=thickness of membrane in structure that use the same carrier. Problem is d = diffusion coefficient – lipophilicity of drug that Isoniazid and VitB6 are formulated together depends on (1) degree of ionization – many and are taken at the same time. Isoniazid can drugs are weak acids/bases. block VitB6 or vice versa. This reduces the o Note that the non-ionized form will pass the efficacy of the drug. This can be solved through membrane. Ionized forms will have less timing. Advise the patient to take isoniazid in the diffusion coefficient, thus lesser degree of morning and Vit B6 in the evening. transport. 3. Saturability – based on a limited number of o For a weak acid: carriers; even if you give higher doses of the The lower the Ka (dissociation drug, the rate of transport will remain the same constant) or higher the pKa (where pKa o First-order kinetics – rate of transport =-log Ka) the faster is the rate of increases as you increase the dose passive transport o Zero – order kinetics – rate of transport o For a weak base: remains the same even if you increase the The higher the Ka or the lower the pKa, concentration the faster is the rate of passive o When a drug at low concentration behaves transport at first-order kinetics, and at high Also depends on partition coefficient (lipid-H2O) concentration behaves at zero-order The dissociation of a weak acid or weak base is kinetics, this is called capacity-limited guided by the Le Chatelier’s Principle: kinetics/ saturable kinetics/MichaelisWeak acid: Menten kinetics – this is characteristic of ALL CARRIER-MEDIATED processes Weak Base: o Team 7 In an acidic environment, with a weak acid, the reaction will shift to the left, towards its non-ionized form (HA). In the acidic C. Convective Transport Transport across pores/channels important for movement of small ions (K+, Na+, etc.) Page 2of4 PRINCIPLS OF PHARMACOKINETICS (1) Year Level 6 [Module 2: General Concepts of Disease and Therapeutics] July 8, 2009 Graph 3. Bioequivalence D. Pinocytosis Vesicle-mediated, energy requiring transport Exception to the rule that the drug must be in aqueous form Drugs that undergo pinocytosis are LARGE LIPIDS The drug must be in the form of MICELLES – lipid globule stabilized by surfactants (surface-active agents i.e., bile salts in the small intestines) Examples: large lipids, fat-soluble vitamins (ADEK) IV. ABSORPTION A. B. Definitions Physiologic definition: rate and extent of disappearance of the drug from the site of administration (or from the site it was administered) Ex. PO (per orem) drug – physiologically absorbed in the gastrointestinal mucosa (entering the portal circulation) Pharmacologic definition: rate and extent of drug entry into the SYSTEMIC CIRCULATION (circulation AFTER the liver; AFTER the portal circulation) o test: extract blood from the peripheral vein and detect the drug in the sample Bioavailability measure of the RATE and EXTENT of drug entry into the systemic circulation Bioavailability studies – subjects are healthy people of same age and weight Bioavailability graph – differs according to whether drug is given intravenously or extravascularly Graph 2. Bioavailability of a Drug Where Cmax- measures both rate and extent – most variable parameter Tmax – measures the rate (how fast the drug is absorbed) AUC – measures extent (how much of the drug is absorbed) – most important parameter (i.e., for BFAD) Note: these three parameters are subject to intra- and inter-individual variability C. Bioequivalence a measure of the similarity in the bioavailabilities of generic and the innovator drug product Team 7 D. Ratio is computed for the three parameters (Cmax, Tmax, AUC) The denominator is always the standard (innovator drug) Two drugs are bioequivalent if the ratio is between 0.8 and 1.25. Bioequivalent studies are only conducted on drugs with bioavailability problems. If the drug is administered directly into the systemic circulation, it is expected, by definition, to have 100% bioavailability Factors affecting Absorption o Dose size administered – the bigger the dose, the higher the absorption (greater rate and absorption) o Ex. Biogesic- 500 mg paracetamol vs Tylenol 650 mg – it is expected that Tylenol would have greater rate and extent of absorption o Surface area of absorption – explains why inhalational drugs have very high absorption rates and extents (lungs have very large surface areas) o Ex. Stomach vs small intestine (SI) – most drugs will be absorbed in the SI because of the villi and microvilli (even for weak acids, surface area of SI is much greater than that of the stomach) o Exceptions: aspirin, ethanol - highly absorbed in the stomach Degree of perfusion of the absorption site o Ex. Stomach (pancreaticoduodenal artery) vs small intestines (arcuate arteries – more perfused) o this is also another explanation for the high absorption in the lungs (alveolar capillaries are abundant) o a MODIFIABLE factor o Lidocaine – local anesthetic for minor operations, can cause problems if systemically absorbed (can cause cardiac arrhythmias) Solution: give lidocaine with epinephrine (a vasoconstrictor) - the blood vessels will constrict, minimizing the systemic absorption of lidocaine Page 3of4 PRINCIPLS OF PHARMACOKINETICS (1) Year Level 6 [Module 2: General Concepts of Disease and Therapeutics] July 8, 2009 o For topical steroids, make sure you instruct the patient not to rub the skin since this will dilate the vessels and cause the drug to enter systemic circulation Gastric emptying time (GET) – important for drugs given per orem (PO) or swallowed drugs o The time it takes for the stomach to empty all of its contents (2-3 hours) o The stomach is a poor absorbing environment because of 1. a small surface area 2. low degree of perfusion 3. lined by thick mucus o Factors that increase GET and delay rate of absorption 1. Hot food, high-protein food, high-fat food 2. Stress, heavy exercise 3. Gastric ulcer 4. Lying on the left side (due to position of the stomach) 5. Anti-motility drugs i.e., opioids (imolium, loperamide), anticholinergic drugs o Factors that decrease GET and increase rate of absorption 1. Cold food 2. Mild exercise 3. Gastrectomy 4. Diabetes Mellitus 5. Lying on the right side 6. Motility enhancers i.e., Metoclopromide (Plasyl), Domperidone (Motilium) FORMAL SPACE FILLERS HO! Bigger resolutions of graphs para sa mga nearsighted: Team 7 Page 4of4