Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Molecular mimicry wikipedia , lookup
Adaptive immune system wikipedia , lookup
Psychoneuroimmunology wikipedia , lookup
Lymphopoiesis wikipedia , lookup
Monoclonal antibody wikipedia , lookup
Polyclonal B cell response wikipedia , lookup
Atherosclerosis wikipedia , lookup
Cancer immunotherapy wikipedia , lookup
Adoptive cell transfer wikipedia , lookup
Immunosuppressive drug wikipedia , lookup
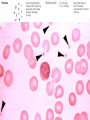
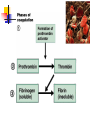
Blood 17 Blood is a viscous suspension: pH 7.35-7.45 Temperature 38C Volume of blood is about 5L (8% body weight) A Liquid Tissue Functions of Blood Distribution: Regulation: O2, nutrients, metabolic products/wastes, hormones, H2O Temp, pH, fluid volume Protection: Fluid/blood loss prevention, prevention of infection Components of Whole Blood Plasma (55% of whole blood) Buffy coat: leukocyctes & platelets (<1% of whole blood) Formed elements 1 Withdraw blood & 2 Centrifuge place in tube Composition: Plasma: about 55% Formed Elements: about 45% Erythrocytes (45% of whole blood) Figure 17.1 Plasma whole blood Cells Plasma Mostly H2O, straw-colored > 100 solutes: electrolytes, gases, hormones, proteins & metabolic products Predominate protein: Albumin (responsible for plasma osmotic pressure) Formed Elements Erythrocytes (RBCs), leukocytes (WBCs), & platelets (PLTs) Only WBCs are complete cells RBCs have no nuclei or organelles Platelets are cell fragments Blood Cell Production Hematopoiesis Tissue: Red bone marrow Flat bones Epiphysis of femur & humerus Cell: all blood cells arise from Hematopoietic Stem Cells (Hemocytoblasts) Erythrocytes (RBCs) Biconcave discs, 7.5 microns Anucleate, essentially no organelles Filled with hemoglobin (Hb) Shape is maintained by a protein framework (spectrin) & other proteins: Figure 17.3 Gives RBCs their shape & flexibility RBC function is gas transport/exchange Erythrocyte Function: Hb 2 a & 2 b chains, each bound to a heme group Heme: Fe containing organic ring structure (porphyrin) Reversibly binds O2 ; each Hb can bind 4 O2 Each RBC has 250 million Hb molecules = 1 billion O2 Hemoglobin Oxyhemoglobin: Hb bound to O2 Deoxyhemoglobin: Hb after O2 diffuses into tissues Carbaminohemoglobin: Hb bound to CO2 CO2 is bound to the polypeptide not to the heme group Carries about 20% of CO2 in the blood Erythropoiesis (RBC Production): Hormonal Control Erythropoietin (EPO) release by the kidneys is triggered by hypoxia (decreased [O2]) Decreased RBC count Decreased oxygen availability Increased tissue demand for oxygen Effect: EPO causes maturation of precursor cells that are in a committed RBC line Erythropoiesis: RBC Line Effect: EPO causes maturation of precursor cells that are in a committed RBC line Erythropoiesis: Iron (Fe) 65% of Fe stored in Hb: Intracellular Fe stored in protein-Fe complexes (ferritin & hemosiderin) in liver & spleen Fe is transported on transferrin (transport protein) Fe obtained in diet; small daily losses Other dietary requirements: B12 & folate (needed for DNA synthesis), other nutrients. Erythrocyte Disorders Anemia: blood has abnormally low O2 carrying capacity (A symptom not a disease itself) Blood O2 levels cannot support normal metabolism Signs: fatigue, paleness, shortness of breath, & chills Anemia Decreased RBCs Decreased Hb Abnormal Hemoglobin Anemia: Decreased RBCs Decreased RBCs: increased destruction/loss or decreased production Hemorrhagic anemia – blood loss Hemolytic anemia – RBC destruction (due to abnormal Hb, infection, trauma etc.) Aplastic anemia – destruction or inhibition of red bone marrow (impacts other formed elements) Anemia: Decreased Hb Iron-deficiency anemia (small pale RBC): decreased HB Inadequate Fe intake Impaired Fe absorption Secondary to hemorrhagic anemia Pernicious anemia (large pale RBC): Deficiency of vitamin B12 Lack of intrinsic factor (required to absorption B12) Treatment - give B12 Anemia: Abnormal Hemoglobin Abnormal Hb: due to genetic defect Thalassemias (Mediterranean ancestry) Absent or faulty globin chain in Hb: Fragile RBCs Thin, delicate, & deficient in Hb Low RBC count Anemia: Abnormal Hemoglobin Sickle-cell anemia: (African ancestry) Abnormal Hb called HbS HbS has a single amino acid substitution in the beta chain Causes RBCs to become sickle-shaped in low O2 situations RBC debris clog capillaries Polycythemia Polycythemia: excess RBCs - increased blood viscosity/sludging Three main polycythemias are: Polycythemia vera Secondary polycythemia (may be due to marrow cancer) (due to erythropoesis) Blood doping Fate & Destruction of RBCs RBCs are anucleate; do not grow, do not make new protein, do not divide Old RBCs become rigid & fragile. The life span of an RBC is 100–120 days Fate & Destruction of RBCs Dying RBCs are engulfed by macrophages in the spleen Heme & globin are separated & the Fe is recycled to ferritin or hemosiderin Heme is degraded to bilirubin (yellow) Bilirubin is secreted by the liver into the intestine as bile The intestines metabolize it into urobilinogen (green) then to stercobilin (brown) Globin portion is metabolized into amino acids & is released into the circulation Life Cycle of Red Blood Cells Figure 17.7 Leukocytes (WBCs): Characteristics Complete cells: contain nucleus , organelles Diapedesis: the ability to move out of the bloodstream & function Circulatory system used to get WBC to the vicinity Cellular/chemical signals prompt diapedesis Amoeboid movement – WBCs travel through tissues to the target area Positive chemotaxis: WBCs follow a chemical trail to the target WBC Function & Classification Function: Phagocytosis, antibody production, waste clean-up, act as chemical “sharpshooters” Classification: Based upon appearance after staining (Wright’s stain) Granulocytes Visible granules on staining – lobed nuclei Neutrophils, eosinophils, & basophils Granules stain specifically (acidic, basic, or both) Larger & usually shorter-lived than RBCs All are phagocytic cells Figure 17.10a-c Neutrophils Multi-lobed nucleus; neutral staining Granules take up both acidic & basic dyes Contain peroxidases, hydrolytic enzymes, & defensins (antibiotic-like proteins) Phagocytize bacteria & some fungi Eosinophils Large bilobed nucleus Red staining (acidophilic) large, coarse, granules containing digestive enzymes Attack parasitic worms by degranulation Phagocytize immune complexes Basophils Large, dark (basophilic) granules containing histamine inflammatory chemical; vasodilator, attracts other WBCs (antihistamines counter this effect) Interact with IgE antibodies (allergies) to promote degranulation Agranulocytes No visible granules, mononuclear Monocytes & lymphocytes Have spherical (lymphocytes) or kidney-shaped (monocytes) nuclei Figure 17.10e,d Monocytes The largest WBCs Active phagocytes; attack viruses Help activate lymphocytes (immune response) In tissues they differentiate into macrophages Lymphocytes Most found in lymphoid tissue (some circulate in the blood) Two types of lymphocytes: T cells – immune system functions B cells give rise to plasma cells, which produce antibodies Summary of Formed Elements Table 17.2 Summary of Formed Elements Table 17.2 Leukocyte Production Leukopoiesis - hormonally stimulated by cytokines: Interleukins Colony-stimulating factors Macrophages & T cells are most important sources of cytokines Leukocyte (WBC) Formation All WBCs originate from hemocytoblasts Hemocytoblasts differentiate into myeloid stem cells & lymphoid stem cells Stem cells mature & differentiate into specific WBC lines Figure 17.11 Leukocytes Disorders: Leukopenia: abnormally low WBC count some drugs & viruses Leukocytosis: WBC count greater than 11,000/mm3 Infection and/or stress Leukemia Leukemia: cancer involving WBCs Leukemias are named according to the abnormal WBCs Myelocytic – involves cells of myeloid lineage Lymphocytic – involves cells of lymphoid lineage Leukocyte (WBC) Formation Figure 17.11 Leukemia (cont) Acute - involves more primitive cells (blasts) & primarily affects children Chronic - more prevalent in older people Leukemia (cont) Bone marrow becomes overwhelmed with the clone cell population. Cancerous cells crowd out all other cell lines Anemia Decrease in all other cell populations Bleeding problems The cancerous WBCs are nonfunctional Death by hemorrhage & overwhelming infections Treatment - radiation, antileukemic drugs, & bone marrow transplants Platelets (PLTs): Characteristics & Function Characteristics: Platelets are not true cells they are fragments of megakaryocytes Small purple spots on Wright’s stain Function: granules contain a large number of chemicals involved in clot formation Platelet Production: regulated by Thrombopoietin Platelet Production Figure 17.12 Hemostasis A series of reactions designed to stop bleeding Three phases: Vascular spasm Platelet plug formation Coagulation (blood clotting) Vascular Spasm Vascular spasm: vasoconstriction in response to smooth muscle injury & chemicals from damaged cells Platelet Plug Formation PLT adhesion: PLTs adhere to collagen exposed by endothelial damage PLT activation (degranulation): releases chemicals Serotonin – enhances vasospasm ADP – attracts more PLTs Thromboxane A2 – enhances serotonin & ADP release (positive feedback) As PLTs aggregate a plug is formed PGI2 (prostacyclin) a prostaglandin made by endothelium limits the extent of the plug Coagulation Thirteen clotting factors (I – XIII) react to transform blood from a liquid to a gel Follow the intrinsic & extrinsic pathways yielding prothrombin activator Final Common Pathway: Final three steps of this series of reactions are: Prothrombin activator is formed Prothrombin is converted into thrombin Thrombin catalyzes the joining of fibrinogen into a fibrin mesh Coagulation: Procoagulants (clotting factors) act as enzymes that catalyze the conversion of other factors Conversion of fibrinogen (a plasma protein) to fibrin (insoluble protein strands) forms a net that traps RBCs & PLTs Coagulation Figure 17.13 Coagulation: Final Common Pathway: Final three steps of these reactions; Prothrombin activator is formed Prothrombin is converted into thrombin Thrombin catalyzes the joining of fibrinogen into a fibrin mesh Clot Retraction & Repair Clot retraction: PLTs release actin & myosin which contract & solidify the clot Repair: Platelet-derived growth factor (PDGF) stimulates repair of blood vessel wall Factors Limiting Clot Growth or Formation Two homeostatic mechanisms prevent clots from becoming large Swift removal of clotting factors Inhibition of activated clotting factors Factors Limiting Clot Growth or Formation Fibrinolysis: dissolving the clot Plasminogen is activated by tissue plasminogen activating factor to form; Plasmin which digests fibrin Clot formation must be limited to the area of injury Dilution: clotting factors must be present in high enough concentration to interact Antithrombin III: inactivates thrombin that is not in association with fibrin Disorders of Hemostasis : Thromboembolytic Conditions Thrombus: Embolus: a clot that develops & persists in an unbroken blood vessel a thrombus broken free & moving in the circulation Embolism: an embolus wedged in a vessel Hemostasis Disorders Disseminated Intravascular Coagulation (DIC): runaway coagulation in intact blood vessels Consumes clotting factors - residual blood cannot clot Untreated: severe hemorrhage Hemostasis Disorders: Bleeding Disorders Thrombocytopenia: insufficient platelets Spontaneous bleeding Petechiae - small bruises Often due to viral infection May be due to medication or bone marrow destruction Liver dysfunction - failure to synthesize procoagulants May be due to vitamin K deficiency Hepatitis/cirrhosis Hemostasis Disorders: Bleeding Disorders Hemophilias: hereditary bleeding disorders Hemophilia A (Classic): factor VIII deficiency Hemophilia B: factor IX deficiency Both A & B are sex linked recessive Hemophilia C: factor XI deficiency, a milder form that can effect both sexes Definitions: Blood antigens Antigen; a substance that is capable of eliciting an immune response Antibody; a protein released by the immune system that binds to a specific antigen Blood Groups Humans have >30 varieties of naturally occurring RBC surface antigens M, N, Duffy, Kell, & Lewis are mainly used for legalities Lansteiner (ABO) group: Corresponding antibodies are naturally present & can cause vigorous transfusion reactions Rh Blood Groups Antibodies to the D (Rh+) antigen are not naturally present If an Rh– individual receives Rh+ blood, anti-Rh antibodies form A second exposure to Rh+ blood will result in a typical transfusion reaction Hemolytic Disease of the Newborn First pregnancy: Rh– Mom & Rh+ infant yields no reaction but, antibodies are formed Second pregnancy: sensitized Rh– mother with Rh+ infant; antibodies cross the placenta & attack & destroy the RBCs the baby The drug RhoGAM can prevent the Rh– mother from becoming sensitized Transfusion Group/type specific blood (i.e. give A+ patient A+ blood): cross match Universal donor (O-) Neither A nor B nor Rh on cells Use only packed cells not O- plasma Autologous transfusion: patient receives their own blood Transfusion Reaction Occurs when recipient plasma has antibodies to donor cells (antigens) Can involve antigens other than A, B or Rh Sx (symptoms): fever, chills, hypotension, tachycardia, nausea/vomiting, etc. Can result in renal failure.