Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
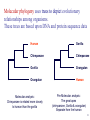
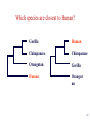
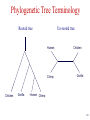
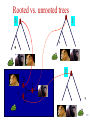
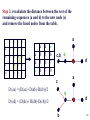
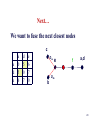
Summary on similarity search or Why do we care about far homologies ? A protein from a new pathogenic bacteria. We have no idea what it does A protein from a model organism. We know its function in one organism but not in another (arrestin) A protein related to a disease -Completely unknown function -May have A different function related to the disease 1 retinol-binding protein apolipoprotein D odorant-binding protein RBP4 and obesity retinol-binding protein apolipoprotein D odorant-binding protein Scoring matrices let you focus on the big (or small) picture PAM250 PAM30 retinol-binding retinol-binding protein protein Blosum80 Blosum45 PSI-BLAST generates scoring matrices more powerful than PAM or BLOSUM retinol-binding protein Phylogenetic trees Phylogeny is the inference of evolutionary relationships. Traditionally, phylogeny relied on the comparison of morphological features between organisms. Today, molecular sequence data are mainly used for phylogenetic analyses. One tree of life A sketch Darwin made soon after returning from his voyage on HMS Beagle (1831–36) showed his thinking about the diversification of species from a single stock (see Figure, overleaf). This branching, extended by the concept of common descent, Phylogeny in Greek =the origin of the tribe 7 Haeckel (1879) Pace (2001) 8 Molecular phylogeny uses trees to depict evolutionary relationships among organisms. These trees are based upon DNA and protein sequence data Human Gorilla Chimpanzee Chimpanzee Gorilla Orangutan Orangutan Human Molecular analysis: Chimpanzee is related more closely to human than the gorilla Pre-Molecular analysis: The great apes (chimpanzee, Gorilla & orangutan) Separate from the human 9 What can we learn from phylogenetics tree? 10 Determine the closest relatives of one organism in which we are interested • Was the extinct quagga more like a zebra or a horse? Which species are closest to Human? Gorilla Human Chimpanzee Chimpanzee Orangutan Gorilla Human Orangut an 12 Human Evolution Neanderthals Modern Man 13 Help to find the relationship between the species and identify new species Example Metagenomics A new field in genomics aims the study the genomes recovered from environmental samples. A powerful tool to access the wealthy biodiversity of native environmental samples 14 Discover new species in the ocean 106 cells/ ml seawater 107 virus particles/ ml seawater >99% uncultivated microbes Discover new species in our own gut The total number of genes in the various species represented in our internal microbial communities (microbiome) likely exceeds the number of our human genes by at least two orders of magnitude. Suez et al, Nature 2014 16 How to discover new species? 17 Extracting Phylogenetic Trees of known species A ? B C D Finding relationships between the unknown and known species 18 Phylogenetic Tree Terminology • Graph composed of nodes & branches • Each branch connects two adjacent nodes R F E A B C D 19 Phylogenetic Tree Terminology Un-rooted tree Rooted tree Human Chimp Chicken Gorilla Chicken Gorilla Human Chimp 20 Rooted vs. unrooted trees 3 3 1 2 1 2 21 How can we build a tree with molecular data? -Trees based on DNA sequence (rRNA) -Trees based on Protein sequences 22 Basic algorithm for constructing a rooted tree Unweighted Pair Group Method using Arithmetic Averages (UPGMA) Assumption: Divergence of sequences is assumed to occur at a constant rate Distance to root is equal Sequence Sequence Sequence Sequence a b c d ACGCGTTGGGCGATGGCAAC ACGCGTTGGGCGACGGTAAT ACGCATTGAATGATGATAAT ACACATTGAGTGTGATAATA a b c d Moving from Similarity to Distance Sequences Sequence Sequence Sequence Sequence a b c d ACGCGTTGGGCGATGGCAAC ACACATTGAGTGTGATCAAC ACACATTGAGTGAGGACAAC ACGCGTTGGGCGACGGTAAT Distances * Dab = 8 Dac = 7 Dad = 5 Dbc = 3 Dbd = 9 Dcd = 8 Distance Table a b c d a 0 8 7 5 b 8 0 3 9 c 7 3 0 8 d 5 9 8 0 * Can be calculated using different distance metrics 24 Constructing a tree starting from a STAR model a a b c d a 0 8 7 5 b 8 0 3 9 c 7 3 0 8 d 5 9 8 0 b d c Step 1:Choose the nodes with the shortest distance and fuse them. 25 Step 2: recalculate the distance between the rest of the remaining sequences (a and d) to the new node (e) and remove the fused nodes from the table. a b c d a 0 8 7 5 b 8 0 3 9 c 7 3 0 d 5 9 8 a 8 a d a 0 5 d 5 0 e 6 7 0 e 6 7 0 c,b e d a c D (ea) = (D(ac)+ D(ab)-D(cb))/2 e d D (ed) = (D(dc)+ D(db)-D(cb))/2 b 26 Step 3: In order to get a tree, un-fuse c and b by calculating their distance to the new node (e) a c a a 0 d 5 e 6 d e 5 6 0 7 7 0 Dce e d Dde b !!!The distances Dce and Dde are calculated assuming constant rate evolution 27 Next… We want to fuse the next closest nodes c a d e a d e 0 5 6 5 0 7 6 7 0 Dce e f a,d Dde b 28 Finally We need to calculate the distance between e and f c f e f 0 4 e 4 0 a Dcee Dde b f Daf Dbf d D (ef) = (D(ea)+ D(ed)-D(ad))/2 29 From a Star to a tree a b f e d b c a d c 30 IMPORTANT !!! •Usually we don’t assume a constant mutation rate and in order to choose the nodes to fuse we have to calculate the relative distance of each node to all other nodes . Neighbor Joining (NJ)- is an algorithm which is suitable to cases when the rate of evolution varies 31 Human Evolution Tree UPGMA Neighbor Joining 32 The down side of phylogenetic trees - Using different regions from a same alignment may produce different trees. Problems with phylogenetic trees 1Bacillus 7Burkholderias 3Pseudomonas 5Aeromonas 6Lechevaliera 2E.coli Salmonella 4 0 .2 Problems with phylogenetic trees Bacillus 1 7 Burkholderias 5 Aeromonas 1Bacillus 3 5 Aeromonas Pseudomonas 3Pseudomonas 7Burkholderias 6 6Lechevaliera Lechevaliera 2 E.coli 4Salmonella 2E.coli 4 Bacillus 3Pseudomonas 7 Burkholderias 5 Aeromonas 6Lechevaliera 2 E.coli 4 Salmonella Salmonella 3Pseudomonas 1 5 Aeromonas 7 Burkholderias 1 Bacillus 6Lechevaliera 2E.coli 4 Salmonella Problems with phylogenetic trees • What to do ? Bootstrapping A.We create new data sets by sampling N positions with replacement. B.We generate 100 - 1000 such pseudo-data sets. C.For each such data set we reconstruct a tree, using the same method. D.We note the agreement between the tree reconstructed from the pseudo-data set to the original tree. Note: we do not change the number of sequences ! 37 Bootstrapped tree Less reliable Branch 1Bacillus 83 58 3Pseudomonas 7Burkholderias 5Aeromonas 6Lechevaliera 2E.coli 100 77 Highly reliable branch 0 .2 4Salmonella Stimulating questions • Do DNA and proteins from the same gene produce different trees ? • Can different genes have different evolutionary history ? 39 40