Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
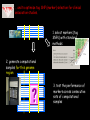
Computational research for medical discovery at Boston College Biology Gabor T. Marth Boston College Department of Biology [email protected] http://clavius.bc.edu/marthlab We study genetic variations because… … they underlie phenotypic differences … cause heritable diseases and determine responses to drugs … allow tracking ancestral human history Our current projects investigate three essential aspects of genetic variations… • how to discover inherited genetic polymorphisms that lead to disease? • how to model human polymorphism structure to inform medical research? • how to select the best genetic markers for clinical case-control association studies? 1. We build computer tools for variation discovery… 1. inherited (germ line) polymorphisms are important as they can predispose to disease the most common type of human polymorphisms are single-nucleotide polymorphisms (SNPs) and short insertion-deletions (INDELs) P( SNP ) all var iable P( S N | RN ) P( S1 | R1 ) ... PPr ior ( S1 ,..., S N ) PPr ior ( S1 ) PPr ior ( S N ) P( SiN | R1 ) P( Si1 | R1 ) S ... PPr ior ( Si1 ,..., SiN ) ... PPr ior ( SiN ) S i1 [ A ,C ,G ,T ] S iN [ A ,C ,G ,T ] PPr ior ( S i1 ) Marth et al. Nature Genetics 1999 we have developed a computer package, PolyBayes© , for accurate discovery of DNA polymorphisms in clonal sequences … we are currently expanding our polymorphism detection capabilities. Homozygous C Heterozygous C/T • for automated detection of somatic single base pair mutations in diploid samples Homozygous T • to include our new knowledge of human variation structure into the detection algorithms • to make the software available for genome centers with high-performance systems and small Biology labs with desktop computers 2. We measure genome-wise distributions of DNA polymorphism data… 0.3 0.2 1. marker density (MD): distribution of number of SNPs in pairs of sequences 0.1 0 0 1 2 3 4 5 6 7 8 9 10 0.1 0.05 0 1 2 “rare” 3 4 5 6 7 8 9 10 “common” 2. allele frequency spectrum (AFS): distribution of SNPs according to allele frequency in a set of samples … we build models of these distributions under competing scenarios of human demographic history… stationary past collapse expansion bottleneck history present MD (simulation) 0.3 0.3 0.3 0.3 0.2 0.2 0.2 0.2 0.1 0.1 0.1 0.1 0 0 0 AFS (direct form) 1 2 3 4 5 6 7 8 9 10 0 0 0 1 2 3 4 5 6 7 8 9 10 0 1 2 3 4 5 6 7 8 9 0 10 0.1 0.1 0.1 0.1 0.05 0.05 0.05 0.05 0 0 1 2 3 4 5 6 7 8 9 10 0 1 2 3 4 5 6 7 8 9 10 1 2 3 4 5 6 7 8 9 10 9 10 0 1 2 3 4 5 6 7 8 9 10 1 2 3 4 5 6 7 8 … and determine the best-fitting models. European data African data genetic bottleneck modest but uninterrupted expansion Marth et al. PNAS 2003; Genetics 2004 3. The HapMap project aims to map out human polymorphism structure to aid gene mapping… However, the variation structure observed in the reference DNA samples genotyped by the HapMap project… … often does not match the structure in another set of samples such as clinical samples used to find disease genes and disease-causing genetic variants … we generate “quasi-samples” with computational means to study sample-to-sample variability… Instead of genotyping additional sets of (clinical) samples with costly experimentation, and comparing the variation structure of these consecutive sets directly… … we generate additional samples with computational means, based on our Population Genetic models of demographic history, using the Coalescent process. … and to optimize tag SNP (marker) selection for clinical association studies. 1. select markers (tag SNPs) with standard methods 2. generate computational samples for this genome region 3. test the performance of markers across consecutive sets of computational samples We are developing projects to expand… • from single-nucleotide DNA changes to developing computer tools for the detection of other types of genomic and epigenetic changes (e.g. in cancer) (Image from Nature Reviews Genetics) • to developing visualization and statistical tools for the integration of diverse genetic and epigenetic data • to using the fruits of the HapMap project, dense SNPs, Linkage Disequilibrium, and haplotype markers to help predict individual responses to drugs, including adverse drug reactions Detecting SNPs in medical re-sequencing data, short insertions / deletions • detection in new data types produced by the latest, super-high throughput sequencing technologies (i.e. 454 Life Sciences sequencing machines) that will be used for individual medical re-sequencing • reliable detection of INDELs and microsatellite polymorphisms, both in clonal and in diploid sequence data, e.g. to detect repeat instabilities Using SNP array data intelligently to detect chromosomal aberrations Speicher & Carter, NRG 2005 Software development for other genetic and epigenetic data (focus on data confidence) copy number detection methylation profile Laird, NRC 2005 chromatin structure Sproul, NRG 2005 Integrate genetic and epigenetic data from varied sources to find “common themes” during cancer development methylation profile chromosome rearrangements chromatin structure copy number changes gene expression profile repeat expansions Using new haplotype resources to connect genotype and clinical outcome in pharmaco-genetic systems • the HapMap was designed as a tool to detect high-frequency (common) phenotypic (e.g. disease-causing) alleles • important drug metabolizing enzymes are relatively few in number, well studied, are at known genome locations, many associated phenotypes are well described • many functional alleles are known, and of high frequency (common) • multi-SNP alleles are highly predictive of metabolic phenotype • clinical phenotype (adverse drug reaction) less predictable • ideal candidate for applying haplotype resources Multi-marker haplotypes as accurate markers for ADRs? genetic marker (haplotype) in genome regions of drug metabolizing enzyme (DME) genes computational prediction based on haplotype structure functional allele (known metabolic polymorphism) clinical endpoint (adverse drug reaction) molecular phenotype (drug concentration measured in blood plasma) Resources • functional alleles • LD and haplotype structure in the HapMap reference samples, based on high-density SNP map • specifics of enzymedrug interactions • existing DME P genotyping chips Evolutionary / PopGen questions • mutations single-origin or recurrent? • geographic origin of mutations? • mutation age? • analysis based on complete local variation structure and haplotype background of functional mutations • specifics of the selection process that led to specific functional alleles? Proposed steps of analysis • complete polymorphic structure? • ethnicity? haplotype block? • additional functional SNPs? • haplotypes vs. functional alleles? • haplotypes vs. metabolic phenotype? • haplotypes vs. ADR phenotype? clinical phenotype (ADR) haplotype functional allele (genotype) metabolic phenotype Funding sources / plans • polymorphism discovery + medical re-sequencing data analysis: 5-year NIH R01 research grant awarded • pop-gen modeling + haplotype analysis + marker selection system: NIH R01 application pending • informatics tools for genomic and epigenetic changes in cancer: need a postdoc to establish project (startup or NIH R21 or private funding) • haplotypes in Pharmacogenomics: need a postdoc to establish project (startup or NIH R21 or private funding)