Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Gene nomenclature wikipedia , lookup
No-SCAR (Scarless Cas9 Assisted Recombineering) Genome Editing wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
Nutriepigenomics wikipedia , lookup
Gene therapy wikipedia , lookup
Population genetics wikipedia , lookup
Gene expression profiling wikipedia , lookup
Epigenetics of human development wikipedia , lookup
History of genetic engineering wikipedia , lookup
Gene therapy of the human retina wikipedia , lookup
Cell-free fetal DNA wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Neocentromere wikipedia , lookup
Y chromosome wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Gene expression programming wikipedia , lookup
Genome evolution wikipedia , lookup
Skewed X-inactivation wikipedia , lookup
Epigenetics of neurodegenerative diseases wikipedia , lookup
Neuronal ceroid lipofuscinosis wikipedia , lookup
Oncogenomics wikipedia , lookup
Helitron (biology) wikipedia , lookup
Designer baby wikipedia , lookup
X-inactivation wikipedia , lookup
Saethre–Chotzen syndrome wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Genome (book) wikipedia , lookup
Haplogroup G-P303 wikipedia , lookup
Frameshift mutation wikipedia , lookup
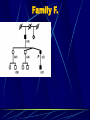
Neurogenetics and DNA laboratory Pavel Seeman CMT team Prague Neurogenetics „neurology of the whole family“ for clinicians : ask about and investigate the relatives ! DNA laboratory in neurological departments at 2nd School of Medicine, Charles University and University Hospital Motol Prague Dept of Child Neurology (Hereditary Neuropathies, Pelizaeus Merzbacher disease, Nijmegen Breakage Syndrome) Dept of Adult Neurology (Hereditary Neuropathies) CMT Dept of Pediatric Otorhinolaryngology (Congenital Nonsyndromic Deafness – Connexin 26) DNA testing for CMT in Czech Rep DNA testing for CMT in the Czech Republic available since 1997 – still the only lab testing for CMT Grants of Ministery of Health of Czech Republic All DNA samples and patient data from CMT patients in one lab – many advantages and great potential Hereditary neuropathies – Charcot-Marie-Tooth diseases Most common group of genetically caused neuromuscular diseases – (1: 2 500 – 5 000) Genetically very heterogeneus group – now mutations in 23 genes can cause CMT Highly prevalent mutation is the CMT1A duplication / HNPP deletion on chromosome 17p incl. PMP22 gene Challenge for the detection methods – to small for cytogenetics, to big for molecular genetics and to close together for conventional metaphase FISH PMP 22 chromosome 17 p 11.2 PMP 22 Normal chromosome 17 p 11.2 PMP 22 chromosome 17 p 11.2 chromosome 17 p 11.2 PMP 22 PMP 22 CMT 1 A PMP 22 chromosome 17 p 11.2 HNPP chromosome 17 p 11.2 PMP 22 chromosome 17 p 11.2 chromosome 17 p 11.2 PMP 22 CMT 1A DSS, CHN HNPP Protein 0 chromosome 1q22q23 chromosome 1q22q23 Protein 0 CMT1B DSS,CHN, CMT2 connexin 32 chromosome X CMTX chromosome X connexin 32 Detection of the most common mutation in CMT in DNA laboratory Set 58°C Set 55°C now: 15 microsatellites markers located within the duplicated CMT1A region Detection of point mutations in further genes in patients with excluded CMT1A/HNPP Connexin 32 gen (GJB1) – in CMTX Myelin protein zero (MPZ) – in severe demyelinating forms and in axonal form PMP22 gene – in demyelinating forms LITAF gene – in demyelinating dominant forms In patients where we have enough clinical data ! Czech CMT families 1997 - 2003 548 - families tested 1271 – individuals mutation detected in total 238 families 140 309 268 individual gene tests 69 CMT1A duplication HNPP deletion Cx32 1 20 8 MPZ PMP22 still unknown CMT1A: 258 individuals, HNPP: 131 individuals CMT families with enough clinical data – 408 families still unknown 42% CMT1A duplicat. 34% in 140 families out of 548 not enough clinical data were provided 408 families with sufficient clinical data HNPP PMP22 MPZ Cx32 5% 0% 2% deletion 17% Modes of inheritance in Czech CMT patients cohort In 392 families – unrelated patients with suficient family information recessive (AR) 1% sporadic 45% dominant: AD + XD 54% Gene testing – sequencing in CMT patients without the CMT1A duplication Connexin 32 gen, GJB1 X- linked dominant expressed in PNS as well as in CNS mutations in Cx32 are the second most common cause of CMT (after CMT1A ) Connexin 32 gene, GJB1 Investigated: 58 families without CMT1A duplication Causal mutation found in 21 families (36,2 %) Among 46 familiar cases only 45,6% Families positive for Cx32 mutation were always large many members affected by CMT One family, possibly a de-novo mutation 6 families from 13 (46%) – carry the same mutation Glu208Lys – haplotype analysis showed a founder efect – all 6 large families have a common founder Mutation Glu208Lys in Cx32 in 6 large Czech families is due to a founder efect. Marker Haplotypes Distance Family K Family B-S Family Z-U Family S-B-V Famiy S-D-H Family S DXS1053 5 2 5 2 1 51.000 kbp DXS8083 5 1 2 2 2 23.000 kbp DXS1111 3 1 1 1 1 2400 kbp DXS453 3 1 1 1 1 1000 kbp DXS8107 1 1 1 1 1 800 kbp GJB1 Glu208Lys Glu208Lys Glu208Lys Glu208Lys Glu208Lys Glu208Lys 0 bp SNP; rs1997625, T>C SNP; rs752081, A>G DXS8060 C C C C C C 1 kbp G G G G G G 5 kbp 4 1 1 1 1 2500 kbp DXS1225 4 1 1 1 1 7000 kbp DXS1197 1 1 1 1 1 8000 kbp DXS8020 2 1 2 1 1 28.000 kbp DXS1206 2 5 1 2 2 54.000 kbp Myelin Protein Zero (MPZ) (P0) gene •expressed in PNS only in myelinated Schwann cells – in central involvement •4 clinical phenotypes – due to a mutation in MPZ – – CMT1 classical (demyel.) - DSS or HMSN III- early onset severe demyel. - congenital hypomyelination (CHN)–very early onset - CMT 2 (axonal) late onset MPZ (P0) gene investigated: 80 families without CMT1A duplication mutation found in 8 families (10 %) 4x axonal form a 4x demyelinating severe form Arg98Cys mutation 2x – in two families de-novo – hotspot PMP22 gene •expression only in PNS – no signs of central involvement •dominant mutations – heterozygotes are affected •phenotype – CMT1 classical - Dejerine Sottas or congenital hypomyelination • PMP22 mutations are rare worldwide PMP 22 gene • all coding exons sequenced in – 33 families/unrelated patients without CMT1A duplication • mutation found in 1 patient only ( 3%) – sporadic case, congenital hypomyelination (CHN) – de-novo mutation Ser72Leu • point mutations in PMP22 are rare LITAF 1/ SIMPLE gene Lipopolysacharide-Induced Tumor necrosis Alfa Factor - Small Integral Mebrane Protein of the Lysosome/late Endosome 3 coding exons, 486 bp, 161 AA Street et al. 2003 – Neurology 60: 22-26 Mutation in LITAF/SIMPLE in CMT1C disease. 46 CMT1 families tested 2 causal mutations detected (>5 %), one is Gly113Ser in a family with very mild CMT Many polymorfisms incl. Ile92Val, Thr78Thr Two different phenotypes caused by P0/MPZ mutation Czech family with axonal CMT beginning with deafness Family F. Family F. hearing loss and deafness as the first symptom of CMT disease - at the late teens in the grandfather and in the mother - progressive hearing loss, deafness now abnormal pupillar reaction before the onset of the neuropathy fully normal physical abilities until the end of 3rd decade late onset of polyneuropathy of axonal type - slow progression at the grandfather but quite fast at the mother, 12 years old boy clinically still unaffected whorsening of electrophysiological and clinical findings correlated with higher age in the family – axonal loss and later demyelination no pes cavus, severe footdrop pronounced hand muscles atrophies Audiograms mother (622) son (623) Electrophysiology Axonal polyneuropathy – very low amplitudes with preserved NCVs Nerve and muscle biopsy in nr. 622 (mother) nerve muscle Normal control 290 A>T (Glu97Val) in MPZ gene Dejerine Sottas neuropathy and MPZ mutation Deafness as late symptom in DSN patients Family K. Mutation Arg98Cys - neighbour aminoacid to the previous family Family K. Mother (44y.) and son (18y.) severely affected (HMSN III or DSN), no other affected members in the family early onset (3 y.) with hypotonia, delayed motor milestones, scoliosis, both affected never achieved normal independent gait distal weakness and atrophies, areflexia, rather nonprogressive course extremely decreased MNCV ( 8-10 m/s), absent SNAP hypertrofic demyelinating neuropathy (with onion- bulbs) in the sural nerve biopsy in the mother in the mother from the age of 25 y. - hearing loss, presently sensorineural hearing loss bilateral ( up to 70 dB) both affected showed abnormal pupillar reaction Family K. mother son Family K. mother no foot deformities son Other, recently discovered genes in CMT (to be screened in the future in „unknown“ patients) PRX - AR demyel GDAP1 - AR - demyel., axonal and intermediar. MTMR2 - AR – focally folded myelin NEFL - AD - axonal and demyel LITAF/SIMPLE - CMT1C - AD, demyel. LMNA - AR - axonal RAB 7 - AD – axonal with ulcers NDRG1 - HMSN Lom - Bulgaria GAN - giant axonal neuropathy NTRK 1 - HSAN IV SPTLC1 - HSN I Larger families with unknown mutation – linkage studies – new candidate genes discoveries HMN II and 12q24 locus CMT196 14 8 20 7 9 10 11 12 13 15 17 16 18 19 23 55 21 22 24 25 26 27 32 1 2 28 31 29 3 30 47 53 48 49 50 51 52 34 33 35 38 40 36 37 39 54 42 41 43 45 44 46 4 5 6 Linkage analysis to localize possible new „CMT genes“ AD CMT family, CMT1A, MPZ excluded Linkage analysis to localize possible new „CMT genes“ AD-CMT family, CMT1A, MPZ, PMP22 mutations excluded electrophysiology intermediate