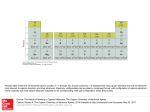
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Fabricating designed fullerene nanostructures for functional electronic devices Christian Larsen Department of Physics Umeå 2014 © Christian Larsen ISBN: 978-91-7601-187-4 Cover art: An almost PCBM transistor Electronic version available at http://umu.diva-portal.org/ Printed by: Print & Media Umeå, Sweden 2014 "Shiny" Abstract A long-term goal within the field of organic electronics has been to develop flexible and functional devices, which can be processed and patterned with low-cost and energy-efficient solution-based methods. This thesis presents a number of functional paths towards the attainment of this goal via the development and demonstration of novel fabrication and patterning methods involving the important organic-semiconductor family termed fullerenes. Fullerenes are soccer-shaped small molecules, with two often-employed examples being the symmetric C60 molecule and its more soluble derivative [6,6]-phenyl-C61-butyric acid methyl ester (PCBM). We show that PCBM can be photochemically transformed into a dimeric state in a bi-excited reaction process, and that the exposed material features a significantly reduced solubility in common solvents as well as an effectively retained electron mobility. This attractive combination of material properties allows for a direct and resist-free lithographic patterning of electronic PCBM films down to a smallest feature size of 1 µm, using a simple and scalable two-step process constituting light exposure and solution development. In a further development, it was shown that the two-step method was useful also in the area-selective transformation of fullerene/conjugated-polymer blend films, as demonstrated through the realization of a functional complementary logic circuit comprising a 5-stage ring oscillator. In another project, we have synthesized highly flexible, single-crystal C60 nanorods with a solution-based self-assembly process termed liquid-liquid interfacial precipitation. The 1-dimensional nanorods can be deposited from their synthesis solution and employed as the active material in field-effect transistor devices. Here, it was revealed that the as-fabricated nanorods can feature an impressive electron mobility of 1.0 cm2 V-1 s-1, which is on par with the performance of a work horse in the transistor field, viz. vacuumdeposited amorphous Si. We further demonstrated that the processability of the nanorods can be improved by a tuned light-exposure treatment, during which the nanorod shell is polymerized while the high-mobility interior bulk is left intact. This has the desired consequence that stabile nanorod dispersions can be prepared in a wide range of solvents, and we anticipate that functional electronic devices based on solution-processable nanorods can be realized in a near future. i ii Sammanfattning Ett stående mål för utvecklingen av organisk elektronik är att kunna realisera flexibla och funktionella elektroniska komponenter genom kostnads- och energieffektiva tillverkningsmetoder. Denna avhandling presenterar möjliga vägar för att kunna uppfylla detta mål genom nya tillverkningsprocesser baserade på en viktig grupp organiska halvledare, fullerenerna. Fullerener består av små fotbollsformade och kolbaserade molekyler där de, inom den organiska elektroniken, mest använda varianterna är C60 och dess lättlösliga derivat [6,6]-phenyl-C61-butyric acid methyl ester (PCBM). Vi demonstrerar att PCBM kan transformeras till dimerer genom en fotokemisk bi-exciterad reaktionsprocess genom enkel ljusexponering. Dimeriserad PCBM visar en attraktiv kombination av egenskaper genom bibehållna elektroniska egenskaper samt en minskad löslighet i vanliga lösningsmedel. Dessa egenskaper möjliggör en direkt och resist-fri mönstringsprocess av det aktiva lagret i organiska fälteffekttransistorer genom att inkludera en tvåstegsprocess bestående av exponering samt framkallning av det aktiva lagret. En mönsterupplösning på 1 µm demonstreras genom mönstring med UV-ljus. Den enkla tvåstegsprocessen kan även användas för att skapa funktionella komplementära kretsar vilket demonstreras genom tillverkning av en 5-stegs ringoscillator. I ett annat projekt tillverkar vi enkristallina, men flexibla, nanostavar av C 60 med hjälp av en uppskalningsbar lösningsmedelsbaserad metod. De 1dimensionella nanostavarna har deponerats och använts som det aktiva materialet i fälteffekttransistorer. Vi visar att dessa nanostavar uppvisar en imponerande elektronmobilitet på 1.0 cm2 V-1 s-1, vilket är jämförbart med vakuum-deponerat amorft kisel som är den nuvarande arbetshästen för kommersiella tunnfilmstransistorer. Vi demonstrerar att processningen av nanostavar kan förenklas genom en ljusexponering som transformerar C 60 i nanostavens skal till en polymerisk struktur, medan stavens kärna förblir intakt. Denna transformation resulterar i att nanostavarna kan användas i ett stort antal lösningsmedel, vilket i sin tur öppnar upp möjligheter för storskalig och lösningsmedelsbaserad tillverkning av funktionella elektroniska komponenter baserat på nanostavar av C60. iii List of Publications I. Direct UV Patterning of Electronically Active Fullerene Films Jia Wang, Christian Larsen, Thomas Wågberg and Ludvig Edman Advanced Functional Materials, 2011. 21(19): p. 3723-3728. http://dx.doi.org/10.1002/adfm.201100568 II. Complementary ring oscillator fabricated via direct laserexposure and solution-processing of a single-layer organic film Christian Larsen, Jia Wang and Ludvig Edman Thin Solid Films, 2012. 520(7): p. 3009-3012. http://dx.doi.org/10.1016/j.tsf.2011.12.048 III. On the fabrication of crystalline C 60 nanorod transistors from solution Christian Larsen, Hamid Reza Barzegar, Florian Nitze, Thomas Wågberg and Ludvig Edman Nanotechnology, 2012. 23(34): p. 344015. http://dx.doi.org/10.1088/0957-4484/23/34/344015 IV. Solution-Based Phototransformation of C60 Nanorods: Towards Improved Electronic Devices Hamid Reza Barzegar, Christian Larsen, Ludvig Edman and Thomas Wågberg Particle & Particle Systems Characterization, 2013. 30(8): p. 715-720. http://dx.doi.org/10.1002/ppsc.201300016 V. Palladium nanocrystals supported on photo-transformed C60 nanorods: Effect of crystal morphology and electron mobility on the electrocatalytic activity towards ethanol oxidation Hamid Reza Barzegar, Guangzhi Hu, Christian Larsen, Xueen Jia, Ludvig Edman and Thomas Wågberg Carbon, 2014. 73(0): p. 34-40. http://dx.doi.org/10.1016/j.carbon.2014.02.028 VI. An Arylene-Vinylene Based Donor-Acceptor-Donor Small Molecule for the Donor Compound in High-Voltage Organic Solar Cells Javed Iqbal, Jenny Enevold, Christian Larsen, Jia Wang, Thomas Wågberg, Bertil Eliasson and Ludvig Edman Submitted iv Publications not included in this thesis: Improving the Performance of Light-Emitting Electrochemical Cells by Optical Design Nikolai Kaihovirta, Christian Larsen and Ludvig Edman ACS Applied Materials & Interfaces, 2014. 6(4): p. 2940-2947. http://dx.doi.org/10.1021/am405530d v List of Acronyms AFM BHJ CV DCFL DSC EDP EQE ETL fcc GND hcp HOMO HRTEM HTL IQE ITO LLIP LUMO MPP NR OFET OLED OPV OSC PCE PDN PDP PNR PUN R2R RFID SAED STEM TCO TEM TGA XRD Atomic force microscopy Bulk heterojunction Cyclic voltammetry Direct-coupled FET logic Differential scanning calorimetry Energy delay product External quantum efficiency Electron transport layer Face-centered cubic Ground Hexagonal close-packed Highest occupied molecular orbital High-resolution transmission electron microscopy Hole transport layer Internal quantum efficiency Indium tin oxide Liquid-liquid interfacial precipitation Lowest unoccupied molecular orbital Maximum power point Nanorod Organic field-effect transistor Organic light-emitting diode Organic photovoltaic Organic semiconductor Power conversion efficiency Pull-down network Power delay product Polymerized nanorod Pull-up network Roll-to-roll Radio-frequency identification Selective area electron diffraction Scanning transmission electron microscopy Transparent conductive oxide Transmission electron microscopy Thermal gravimetric analysis X-ray diffraction vi Table of Contents 1. Introduction 1 2. Organic semiconductors 2 2.1. The fullerenes 4 3. Electronic devices 6 3.1. The field-effect transistor 7 3.1.1. The inverter 11 3.1.2. The ring oscillator 15 3.2. The photovoltaic cell 16 3.2.1. The bilayer structure and operation principles 17 3.2.2. The bulk heterojunction solar cell 20 3.2.3. The OPV performance 21 4. Fullerene photochemical transformation 24 4.1. Raman spectroscopy 24 4.2. Fullerene patterning by resist-free lithography 25 4.3. Patterning efficiency 27 4.4. The photochemical reaction mechanism 29 4.5. PCBM OFETs 31 4.6. Realizing complementary logic circuits with resist-free patterning 33 4.6.1. The inverter 34 4.6.2. The ring oscillator 36 5. Fullerene C60 nanorods 38 5.1. Synthesis 38 5.1.1. Polymerization 40 5.2. Characterization 41 5.2.1. As-grown 41 5.2.2. Annealing 43 5.2.3. Polymerization 44 5.3. The C60 nanorod transistor 46 5.3.1. Nanorod deposition 47 5.3.2. Transistor performance 49 6. Material design for improved photovoltaics 52 6.1. A small-molecule donor compound for OPVs 52 6.2. The C60 nanorod OPV 56 7. Conclusions 58 Acknowledgements 59 References 61 vii viii 1. Introduction The field of organic electronics includes the design, synthesis, characterization and application of organic compounds with electronic properties. These organic compounds are small molecules or polymers with a conjugated, carbon based, backbone. Since carbon is an abundant material on earth, these materials promise to be produced and recycled inexpensively at a very large scale. The chemical tailoring possibilities of conjugated materials also allow for synthesis of materials with a wide variety of properties, making them suitable for both electronic applications (such as transparent, large surface-area electrodes and transistors) and photonic applications (such as light-emitting devices and solar cells) [1-4]. Conjugated materials can also be tailored for improved processability in e.g. lowtemperature solution processes. This subsequently allows the use of unconventional flexible substrates, such as plastic and paper, which are compatible with roll-to-roll fabrication, allowing for high throughput device fabrication [5-9]. Organic conjugated materials are already used in commercial applications, with the most noteworthy being the organic lightemitting diode (OLED) displays. Possibilities for future applications include intelligent packaging, flexible displays [10], radio-frequency identification (RFID) tags [11], transparent or paintable solar cells [12] and light-emitting wallpaper [5]. This thesis is structured such that the basics of organic semiconductors (OSCs) are covered in chapter 2. In chapter 3 the device physics of organic field-effect transistors (OFETs) and organic photovoltaic (OPV) cells are disclosed. Chapter 4 describes the process of photochemical transformation of fullerene molecules to create dimers/oligomers/polymers, and how this transformation can be used to realize efficient electronic devices and complementary logic circuits. Chapter 5 presents a solution-based method for the synthesis of crystalline C60 nanorods, and describes how such nanostructures can be practically implemented as the active material in OFETs. Finally, chapter 6 presents an innovative solution to the detrimental issue of fullerene aggregation in OPVs in the form of a morphology-locked active material based on C60 nanorods. 1 2. Organic semiconductors Figure 1. (a) The electron orbitals of a sp2 hybridized carbon atom, with the three sp2 orbitals positioned at a 120° angle with respect to each other, and the 2pz orbital positioned in a direction perpendicular to the plane spanned by the sp2 orbitals. (b) The overlap of two 2pz orbitals on neighboring carbon atoms in a conjugated molecule will give rise to π-orbitals (red), as shown for ethene. Organic conjugated materials comprise a molecular backbone of carbon atoms connected through alternating single and double bonds in a planar configuration. The planarity originates from the sp2 hybridization, where the 2s, 2px and 2py orbitals of the carbon atom hybridize into three degenerate in-plane sp2 orbitals, positioned at a 120° angle with respect to each other. The 2pz-orbital of the carbon atom is positioned in a perpendicular direction to the plane spanned by the three sp2 orbitals, as shown in figure 1(a). Strong covalent σ-bonds are formed between two neighboring sp2 hybridized carbon atoms through the overlap of two neighboring sp2 orbitals. The two neighboring pz orbitals will also overlap to give rise to a new π-orbital, which contains delocalized π-electrons. These π-orbitals provide the rigidity of a conjugated molecule, since they need to be broken in order to rotate the molecule. The σ- and π-bonding of an ethene molecule (C2H4) is shown in figure 1(b). For a molecule in the ground state, the most energetic electron is positioned in the highest occupied molecular orbital (HOMO). The next available electron state is termed the lowest unoccupied molecular orbital (LUMO), and the energetic difference between the LUMO and HOMO levels is termed the energy gap (Eg). In long-chain conjugated molecules, the most energetic 2 π-electrons can delocalize over several repeat units, and the length of this delocalization is called the conjugation length. By increasing the conjugation length, the energy gap between the LUMO and HOMO levels will decrease in a monotonic fashion, as shown schematically for an increasing number of connected thiophene monomers in figure 2 [13]. During this process, the LUMO+1, LUMO+2 levels, etc. can become so close in energy that it, in some cases, make sense to term them a LUMO band or conduction band; the same argumentation for the HOMO levels motivates the formation of a HOMO band or valence band. As the ground-state configuration (at zero Kelvin) for a conjugated compound is a filled HOMO band and an empty LUMO band, these materials are appropriately classified as semiconductors. Figure 2. Electronic structure as a function of number of thiophene rings. At sufficiently long conjugation length, the discrete energy levels become degenerate and form a band-like structure. An organic semiconductor (OSC) can be reduced (oxidized) through the addition of an electron (hole) into the LUMO (HOMO) band. Once injected, the electron is free to move within the π-states, and this intra-molecular transport is commonly facile [14]. An inter-molecular transfer step is however necessary in order for the electrons to propagate beyond the conjugation length, and this is a comparatively slow process. The ease with which charge carriers move is quantified by the mobility (µ, unit: cm2 V-1 s-1), which can be calculated as the drift velocity of the charge carrier (v, unit: cm s-1) divided by the driving electric field (E, unit: V cm-1). 3 When OSC molecules are in close proximity of each other, as in a solid film, the π-orbitals of different molecules can overlap. This overlap can result in a more efficient inter-molecular charge transport, and molecular ordering, through crystallization, can increase the molecular π-orbital overlap and the mobility significantly [15, 16]. The conductivity (σ) of a semiconductor is given by the equation qnn qpp (1) where n and µn are the concentration and mobility of the electrons and p and µp are the concentration and mobility of holes. 2.1. The fullerenes An exceptionally interesting type of organic semiconductor is the fullerene. A fullerene comprises at least 20 carbon atoms arranged in hexagonal and pentagonal rings (the smallest fullerene C20 only comprises pentagonal rings), and it can form spherical, cylindrical, ellipsoidal and many other shapes. The most common fullerene is C60, which is built up from 60 carbon atoms in alternating hexagonal and pentagonal rings to form a shape similar to a football, as shown in figure 3. Figure 3. The C60 fullerene (left) with alternating hexagonal and pentagonal rings of conjugated carbon. The shape can be directly compared to a football (right). C60 and several of its derivatives are excellent organic semiconductors, with some rather unique features, such as high electron mobility and low LUMO level. They have therefore been used extensively as the active material in OFETs, and as the electron acceptor material in OPV devices. The symmetric form of the spherical C60 molecule makes it prone to crystallization. 4 Moreover, the π-orbitals of C60 are positioned around the entire molecule and inter-molecular π-orbital overlaps are accordingly possible in all directions. This combination of properties is the origin to why C60 features one of the highest values for the electron mobility among OSCs [17]. Additionally, different C60 molecules can be made to chemically connect in a photo-chemical process, so that dimers/oligomers/polymers can be created [18]. One drawback with C60 is that its solubility in organic solvents is limited, making thermal evaporation the deposition method of choice. This deposition method results in high quality films, but suffers from being very time-consuming and expensive as compared to solution-based processes. The solubility issue can however be resolved through chemical tailoring. By adding a side chain to the C60 molecule to produce the derivative [6,6]phenyl-C61-butyric acid methyl ester (PCBM), the solubility is drastically improved. 5 3. Electronic devices Figure 4. The basic structure for electronic devices consisting of two electrodes connected by a thin film of an organic semiconductor in a planar (a) or sandwich (b) configuration. Since their discovery, OSC materials have been introduced into a wide variety of applications, notably transistors, solar cells and light-emitting devices. Although the operational mechanisms differ, these devices share a common feature in that they comprise a thin OSC film contacted by two electrodes. The electrode configuration can be either planar (figure 4(a)), with the current flowing along the substrate, or vertical or sandwich (figure 4(b)), with the current flowing in a direction perpendicular to the substrate. A schematic energy level diagram for the constituent components of a 2electrode structure at open circuit is shown in figure 5(a). As intrinsic organic semiconductors by definition are undoped, efficient charge injection can be achieved by minimizing the injection barrier (qΦ) at the electrode/OSC interface. For electron injection, this injection barrier corresponds to the energy difference between the cathode work function (EC) and the OSC LUMO level (ELUMO), and for hole injection it corresponds to the mismatch between the anode work function and the OSC HOMO level (EHOMO). In figures 5(a-c) the electrodes were chosen such that the left electrode, the cathode, has a low work function, hence favoring electron injection, while the right electrode, the anode, has a high work function that favors hole injection. 6 Figure 5. Schematic energy level diagram of a 2-electrode device structure at (a) open circuit, (b) short circuit, (c) during ambipolar hole and electron injection, and (d) during unipolar electron injection. The use of electrodes with different work functions introduces an asymmetry into the device, which results in a built-in-field (Vbi) in the active material under short-circuit conditions, as indicated in figure 5(b). By applying a forward bias (VF) larger than the Vbi, the injected charges will be driven by the electric field towards the opposing electrode, as seen in figure 5(c). If the device is biased in the reverse direction, the injection of electrons and holes will be severely limited by the large injection barriers. The described device is thus a diode, which rectifies the current in the forward direction [19-21]. If one instead uses the same material for both electrodes, it is possible to achieve efficient injection of only one carrier type, and the operation of an electron-only device is depicted in figure 5(d). 3.1. The field-effect transistor The realization of the transistor and the integrated circuit sparked a technical revolution, which has resulted in faster, smaller and more efficient electronic devices. Following the demonstration of the metal oxide semiconductor field-effect transistor (MOSFET), the efficiency of the integrated circuits was further increased, eventually leading up to the high- 7 performance electronic tools and toys we have around us today. However, the processing of the constituent inorganic semiconductor materials, most commonly Si, is expensive, energy consuming, and executed at high temperatures not compatible with flexible substrates. OSCs, on the other hand, can due to their solubility and intrinsic flexibility offer a complementary category of low-cost and flexible electronic devices, which is not available for their inorganic counterparts. OFETs are fabricated in a planar geometry, with the electrodes in contact with the OSC termed source and drain. The OFET structure also comprises a third gate electrode separated from the OSC and the source and drain by an insulating dielectric layer [22-25]. The role of the gate is to capacitively induce charges in the OSC channel next to the dielectric through the application of a gate voltage. The OSC conductivity is thus controlled by the gate voltage, as described by equation (1). Figure 6. Schematic representation of the four different OFET geometries, with the definition of the gate length, LG, and the drain width, WD, included in the bottom-gate, bottom-contact OFET in (a). There are four OFET geometries, which are distinguished by the position of the active OSC layer and the electrodes, as schematically depicted in figure 6. The bottom-gate (BG) geometry has the gate electrode located directly on the substrate and below the active layer1, whereas the top-gate (TG) geometry has the active material directly on the substrate with the gate on top. 1 The BG geometry is commonly used in research since doped Si with a thermally grown SiO 2 layer is a substrate which simultaneously fulfills the role as both gate electrode and dielectric layer, making fabrication procedures simple. 8 Furthermore, the source and drain electrodes can be either in top-contact (TC) or in bottom-contact (BC) depending on whether they are deposited on top or below the active material. The schematic figure of the BG-BC geometry, shown in figure 6(a), also includes the definitions of the gate length (LG) and the drain width (WD). Figure 7. Schematic presenting the operation of an OFET (a) in the linear region, (b) at the pinch-off point and (c) in the saturation region. The dark red color indicates the charge accumulation in the channel. (d) Typical output data with the different regions indicated. The OFET operation can be separated into three regimes: the sub-threshold region, the linear region and the saturation region. The threshold voltage (VT) is defined as the gate-source voltage (VGS) at which all immobile trap states in the OSC channel are filled. Hence, in the sub-threshold region, when VGS < VT, effectively no mobile charge carriers exist in the OSC channel and only leakage currents can be measured. When VGS > VT, mobile charge carriers are induced in the channel and the current through the device is greatly increased. In the linear regime, when VDS < VGS - VT, the potential in the entire channel (from source to drain) is above VT, and the drain-source 9 current (IDS) is increasing linearly with increasing VDS, as shown in figure 7(a). When VDS = VGS - VT the channel potential at the drain electrode equals VT, and this is the pinch-off point, as shown in figure 7(b). When VDS > VGS VT, the device operates in the saturation region, as shown in figure 7(c). In this region the IDS is limited by the amount of charges injected into the channel from the source electrode and is hence dependent on VGS - VT [26]. The IDS in the linear and saturation regimes can derived by the long-channel approximation, and the corresponding analytical expressions are [27-29]: I DS, l i n I DS, sa t C GWD LG C GWD 2 LG V2 VGS VT VDS DS , VGS > VT ; VDS ≤ VGS - VT 2 (2) VGS VT 2 , V (3) GS > VT ; VDS ≥ VGS - VT Here, CG is the gate capacitance, which depends on the permittivity and thickness of the dielectric layer, and µ is the charge-carrier mobility. Note that the mobile charge-carrier in the OFET channel can either be a hole or an electron, and that the OFET accordingly is termed p-type or n-type. In some cases, both n-type and p-type operation can be measured on the same OFET device, and such OFETs are termed ambipolar. Figure 8. (a) 3D representation of OFET characteristics, with the output data indicated by black lines and the transfer data indicated by grey lines. (b) The square root of IDS as a function of VGS, with which it is possible to extract VT and the mobility, µ, through the relationship in equation (3). Typical OFET characteristics are shown in the 3D plot in figure 8(a), with the output data indicated by the black lines and the transfer data indicated by 10 the grey lines. There are a several ways to determine the charge carrier mobility of the OSC with an OFET, but perhaps the most straightforward method is to plot the square root of IDS as a function of VGS at saturation conditions, i.e. at VDS > VGS - VT. By applying a linear fit (solid line in figure 8(b)) it is possible to determine VT as the voltage where the extrapolation of the linear fit crosses the x-axis (i.e. when IDS = 0). With the aid of equation (3) and information on the geometrical parameters CG, WD and LG, it is then possible to also determine the charge carrier mobility [29]. 3.1.1. The inverter The inverter, which is designed to invert an incoming signal, is based on transistors and is the simplest digital logic element in integrated circuits. It is therefore used extensively to show that a new transistor technology is viable for integration into digital integrated circuits. Several inverter designs exist, and the most suitable for a given transistor technology is set by the properties of the specific constituent transistors. Figure 9. Three schematic alternatives to realize an inverter: (a) a resistive PUN design, (b) a DCFL design and (c) the superior complementary design. The simplest design is using resistors in a pull-up network (PUN), i.e. the part of the circuit responsible for pulling the output high (VDD), and an ntype transistor in a pull-down network (PDN), i.e. the part of the circuit responsible for pulling the output low (GND, ground), as show in figure 9(a). A careful selection of RPUN is important in order to achieve fast and robust operation. It is however notable that this design will always suffer from a high supply current (IDD) at high input voltage (Vin), and consequently a low output voltage (Vout), due to the direct current path between VDD and GND when the n-FETPDN is on. The requirements on RPUN in order to achieve robust operation will also result in a slow low-to-high Vout switching speed. 11 Another option using only n-type transistors is the direct-coupled FET logic design (DCFL), where the PUN comprises an FET with the gate and drain connected2, as shown in figure 9(b). In this design the n-FETPUN will operate similar to a diode, and a careful design of the WD for both the n-FETPUN and the n-FETPDN is important to achieve fast and robust operation. Due to the nonlinear behavior of the PUN in this design it is possible to achieve faster switching speeds, but the design will still suffer from a direct current path for IDD at high Vin. Both designs can also, with minor changes, be realized using p-type FETs [30]. Figure 10. Inverter stability diagram showing Vout (solid line), its inverse (dotted line), and IDD (dashed line) as a function of Vin. The different operation regimes for the constituent OFETs are indicated and marked with the open circles and the letters (a)-(e). If a transistor technology can integrate both n- and p-type FETs into an integrated circuit then it is possible to achieve the complementary inverter design, as shown in the schematic in figure 9(c) [31]. In this design the PUN comprises a p-type OFET, while the PDN comprises an n-type OFET. This design is the superior alternative, since only one FET at a time is conducting 2 It is also possible to connect gate and source in the PUN, although this requires a transistor with negative VT in order to operate at a decent speed. 12 during static operation. This greatly reduces the power consumption of the inverter circuit, since only leakage currents through FETs operated in the sub-threshold region will be dissipated during steady-state operation. The principles of static operation of a balanced complementary inverter (see figure 9(c)) is presented in the stability diagram in figure 10, which includes Vout (solid line) and IDD (dashed line) as a function of Vin. The shape of the curves is dictated by the operation regimes of the constituent OFETs 3: (a) When Vin is low, the p-FET operates in the linear regime, while the n-FET is in the sub-threshold region, i.e. off. At this point IDD is limited by the low nFET sub-threshold current. (b) With increasing Vin, the VGS for the n-FET will eventually reach and exceed VT. This corresponds to the entering of its saturation regime, and the point where the channel of the n-FET starts to become conducting. At this point Vout will begin to decrease and IDD will start to increase, since both FETs are now entering an on-state operation regime. (c) By further increasing Vin, the p-FET is also pushed into saturation, thus further increasing the IDD and allowing a very rapid change in Vout. The slope of the stability curve, denoted as the gain, reaches a maximum at this point. This peak value is an interesting metric of the inverter as it describes how sharp the switching from low to high (and vice versa) is. (d-e) A further increase in Vin will move the FETs through the reverse behavior, as observed before the switching threshold. A functional inverter thus features a high Vout when Vin is low, and vice versa, as well as a sharp switching threshold which is quantified by the gain, g = dVout/dVinmax. An indication of the robustness of the inverter is obtained by plotting the inverse of Vout (dotted line). A large area between Vout and its inverse is preferable, since this indicates high robustness against noise caused by e.g. interconnections and device-to-device variations [32]. The description so far have only concerned the steady-state operation of the inverter. In order to evaluate the dynamic behavior it is important to consider the charging of capacitances (both intrinsic and parasitic), when charge carriers move through the device. As a first-order approximation it is common to sum up all capacitive elements into a fixed single-load capacitance between the output node and ground (CL), as shown in figure 11(a). The capacitance originating from the overlap between the input and output electrodes is known as a Miller capacitance and should be counted twice in CL [32]. 3 This description assumes that the n-FET have a positive VT and that the p-FET have a negative VT which is the most common case for undoped OFETs. 13 Figure 11. (a) Circuit model describing the dynamic behavior of a complementary inverter and (b) the corresponding dynamic Vout response following a step change in Vin. The dynamic operation of the inverter can be pictured as the charging and discharging of the load capacitance, and the temporal response of Vout to a step in Vin for a balanced complementary inverter (see figure 10) is plotted in figure 11(b). First, the inverter input is held at Vin = GND (= low), so that Vout is high and CL charged. Following a Vin step from GND to VDD (i.e. low-tohigh), the n-FETPDN is forced to open and the p-FETPUN forced to close. This opens up a path to ground through the n-FETPDN for the charge stored in CL, and this dissipation will begin to lower Vout. The time it takes for Vout to reach VDD/2 is defined as the high-to-low propagation delay (tpHL). Eventually all CL charges will be dissipated, and Vout has now reached the low output. When Vin is switched back to GND, the n-FETPDN closes and the p-FETPUN opens, resulting in a charge flow through the open p-FETPUN so that CL becomes charged and Vout increase. The time for Vout to reach VDD/2 is defined as the low-to-high propagation delay (tpLH). The total propagation delay (tp) is defined as the mean of the high-to-low and low-to-high delays, and can be estimated with the following equation: tp 1 tpHL tpLH 1 C LVDD C LVDD 2 2 I DS,PDN I DS,PUN (4) where IDS,PDN and IDS,PUN is measured at |VGS| = |VDS| = VDD for the n-FETPDN and the p-FETPUN, respectively. It is usually desirable to have inverters with similar values for tpHL and tpLH, and according to (4) this can be achieved by balancing the IDS,PDN and IDS,PUN of the constituent OFETs e.g. by varying their WD [32]. 14 The average power consumption (Pav) is another important metric for an inverter technology and it comprises three contributions: Pav Pdyn Pdp Pstat (5) where Pdyn is the dynamic power consumption, Pdp the direct-path power consumption and Pstat the static power consumption. The Pdyn contribution is due to the power dissipated through charging and discharging of CL, and is hence dependent upon the frequency of switching. Pdp originates from the direct path current which arises if both the n-FETPDN and the p-FETPUN are on at the same time during a dynamic switching event. This contribution also depends on the switching frequency. However, this effect can be reduced by increasing the size of CL [32]. Pstat corresponds to the steady-state leakage of the inverters. This contribution is significant for organic FETs, due to their characteristic high sub-threshold currents [33]. 3.1.2. The ring oscillator Figure 12. (a) Schematic showing a 5-stage (N=5) ring oscillator and (b) typical data based on a 5-stage complementary ring oscillator with Vout (thick line) and IDD (thin line). A simple circuit can be realized by connecting an odd number (N) of inverters in series, with the output of the last inverter connected to the input of the first, as shown in figure 12(a) [34, 35]. Due to the odd number of inverters, this circuit will have no stable operation point, and a voltage transition will propagate through the circuit and looped as fast as the inverters allow. The voltage at each output node will thus oscillate with a characteristic frequency. This circuit is hence referred to as a ring oscillator. Due to its simplicity, the ring oscillator is commonly used to evaluate the performance and functionality of the inverter technology. A successful ring 15 oscillator operation requires that each inverter is able to switch and drive the subsequent inverter in the chain. With a successful ring oscillator at hand, the realization of larger logic circuits is often simply a matter of circuit design [36-39]. A typical measurement on a complementary ring oscillator is plotted in figure 12(b), and it presents Vout from one of the output nodes (thick line) and the supply current (IDD) consumed by the oscillator (thin line) as a function of time. The time period (T) for a full oscillation of Vout can be estimated as N × 2tp. The frequency of the oscillation (f) is the inverse of T. It is also possible to determine Pav for an inverter in the ring oscillator by integrating IDD over one time period: T Pa v VDD I DD t dt NT 0 (6) By multiplying Pav with tp the power delay product (PDP) is obtained. The PDP represents a measure of the average energy consumed per switching event. By subsequently multiplying the PDP with tp the energy delay product (EDP) is obtained, and the EDP thus represents a combined energy and performance metric. These metrics allow for a comparison of different inverter technologies, as they describe the performance and efficiency of each technology in a quantitative manner [32, 40]. 3.2. The photovoltaic cell Our modern lifestyle consumes an increasing amount of energy each year. Historically, this energy has been almost exclusively supplied through fossil fuels (coal, oil and gas), a non-renewable energy resource. Along with the problem of exhausting earth's fossil energy resources comes the release of the greenhouse gases (CO2, CH4, etc.), the inevitable product from fossil fuel combustion which, once in the atmosphere, contributes to global warming. Renewable energy alternatives are thus necessary in order to alleviate these problems. One such alternative is the photovoltaic (PV) cell in which solar energy is converted to electrical energy. The current generation of commercial PV cells comprises inorganic Si, and such cells can feature a high efficiency thanks to the maturity of the Si technology. The refining and processing of the Si is however an expensive process, both in terms of price and energy. In 1992, an ultra-fast photo-induced electron transfer from a conjugated polymer to C60 fullerene molecule was demonstrated [2]. This discovery of 16 efficient charge separation in organic materials started the development of a new generation of light-energy-harvesting devices, organic photovoltaics (OPVs), which offer a low-cost and easy-to-fabricate alternative to the existing Si-based PVs [3]. 3.2.1. The bilayer structure and operation principles Figure 13. The (a) device structure and (b) energy diagram for a bi-layer organic solar cell. In order to understand the operation principles of an OPV cell it is educational to start with the simple bi-layer structure. This structure is depicted in figure 13(a) and comprises two organic semiconductor layers sandwiched between two electrodes, of which at least one needs to be transparent. Transparent conductive oxides (TCO), such as indium tin oxide (ITO), is usually used as the transparent electrode, although significant efforts are put into finding replacements based on more abundant, low-cost and flexible materials [41-43]. A thin film of a donor (D) material is first deposited onto the TCO. The role of the donor is to absorb the incoming photons to excite electrons from the HOMO into the LUMO to create excitons, viz. bound electron and hole pairs. An effective donor material should thus feature a high absorption coefficient (~105 cm-1) to allow for complete light absorption for a film with a thickness on the order of 100 nm. Since the exciton is a neutral particle it will diffuse randomly in the donor, independent on the electric field, until it either recombines or dissociates into a free electron and hole. Due to the low permittivity of organic materials, the binding energy of the exciton (Eexciton > 17 100 meV) is much larger than the average phonon energy at room temperature (~25 meV), implying that the exciton will recombine unless a driving force will cause it to dissociate [44]. Figure 14. Schematic highlighting the desired (green solid arrows) and non-desired (black dashed arrows) processes related to (a) photon absorption, (b) exciton dissociation, (c) charge transport and (d) charge collection in an OPV at short-circuit condition. 18 A driving force for exciton dissociation can however be an interface with an acceptor (A) material with lower ELUMO and EHOMO than the donor, so that the electron, but not the hole, finds a more favorable state in the acceptor. The bound exciton can thus dissociate into a free electron located in the acceptor LUMO and a free hole located in the donor HOMO at the D/A interface. Once separated, the charges are free to move in an external field. By selecting a top electrode material with a lower work function than the TCO, it is possible to create an internal electric field which will act as a driving force on the electrons and holes towards the top cathode and the bottom TCO anode, respectively. The successful collection of the charge carriers at the respective electrode results in a PV current. A schematic energy diagram for a functional bi-layer solar cell is shown in figure 13(b). In order to maximize the performance of an OPV it is important to understand and minimize the loss factors. The efficiency with which the OPV converts incoming photons into current exiting the device is given by the equation for the external quantum efficiency (EQE): EQE a bs IQE a bs ed ct cc (7) The internal quantum efficiency (IQE) refers to the efficiency with which the absorbed photons, i.e. those photons that are not reflected or transmitted out of the cell, can generate current. ηabs describes the efficiency of absorption, and this process is presented in figure 14(a), where the green solid arrow indicates the desired events (in this case photon absorption to create an exciton) and the black dashed arrow indicates the non-desired event (a nonabsorbed photon reflected from or transmitted through the device). ηed represents the exciton dissociation efficiency, as schematically shown in figure 14(b). Once created through absorption, the exciton will diffuse in the organic semiconductor until it is either separated into free charge carriers at an interface (green solid arrow) or recombined under the generation of heat or light (black dashed arrow). ηct describes the charge transport efficiency, as depicted schematically in figure 14(c). Once separated, the free charge carriers need to be transported to the correct electrodes in order to be collected (green solid arrows). This process requires a driving force from an electric field or a charge concentration gradient, as well as uninterrupted pathways for the charge carriers. The free electrons and holes can, however, also recombine at a D/A interface before reaching the electrodes (black dashed arrow). ηcc is the charge collection efficiency at the electrodes, as described in figure 14(d), where the non-preferred process is charge collection at the wrong electrode. 19 For bi-layer devices, the good news is that ηabs is efficient at a film thickness of ~100 nm, ηct can be high due to the good mobility in the pure donor and acceptor materials, and ηcc is close to unity since the electrodes are only contacting materials carrying a single charge-carrier type. ηed is, however, greatly limited by the short diffusion length (~10-20 nm) of excitons in OSCs [45]. As a result, only photons absorbed within an exciton diffusion length away from a D/A interface will contribute to the total EQE, thus greatly limiting the total current produced in a bi-layer device. 3.2.2. The bulk heterojunction solar cell Figure 15. (a) A schematic of the BHJ-OPV device structure, with ETL and HTL layers, and (b) the corresponding energy diagram. The bulk heterojunction (BHJ) OPV cell is designed to alleviate the problem of inefficient exciton dissociation. In a BHJ device the donor and acceptor are intimately mixed in a single active-layer film. In such a film, the donor and acceptor materials are able to form interfaces on length scales of only a few nm, i.e. well below the exciton diffusion length, which allows for a high ηed. However, with a randomly intermixed material network come charge transport and collection problems. Since both donor and acceptor materials are in contact with the electrodes in a BHJ device, it is important to ensure that only the correct type of charge carrier is collected at the electrodes. Therefore, a hole-transport layer (HTL), designed to block electrons while efficiently transporting holes, and an electron-transport layer (ETL), designed to block holes while transporting electrons, can be deposited in 20 between the active layer and the anode and the cathode, respectively. The inclusion of a HTL and an ETL minimizes charge collection losses, i.e. keeps ηcc close to unity. A typical device structure for a BHJ-OPV cell is shown in figure 15(a) and the accompanying energy diagram is shown in figure 15(b). Maximizing the charge transport efficiency ηct in a BHJ cell is a delicate matter. In an intimately mixed network the interface surface area is greatly increased, resulting in increased exciton dissociation but also higher recombination probability [46, 47]. The D/A random network (as shown in figure 15(a)) formed during thin-film deposition can also comprise onephase material islands, resulting in interrupted pathways for charge transport. This random D/A network is however not permanent and it can be adjusted after deposition in order to optimize the film morphology. Methods for optimizing the film morphology include thermal annealing [48-50], solvent treatment [51] and light exposure [52, 53]. Still, the overall aim is the same, i.e. controlling the phase separation to reduce charge recombination while maintaining efficient exciton dissociation. 3.2.3. The OPV performance Figure 16. (a) Energy diagram highlighting parameters vital to the OPV cell performance. (b) The AM1.5G irradiance spectrum showing the spectral irradiance as a function of wavelength and energy. The total irradiance is 1 kW m-2. Various material and device parameters determine the maximum performance of an OPV cell, with the energy levels of the D/A materials playing an important role. The maximum voltage is determined by the difference in energy between the acceptor LUMO and donor HOMO, Emax = 21 ELUMO,A - EHOMO,D, as this represents the energy difference between the separated electrons and holes, as seen in figure 16(a). To maximize the voltage, the EHOMO,D is increased and/or the ELUMO,A is decreased. In order to allow for efficient exciton dissociation, and therefore a high current density, the driving force for exciton dissociation, ΔE = ELUMO,D - ELUMO,A, must be larger than Eexciton. Moreover, a lower donor energy gap (Eg = ELUMO,D EHOMO,D) results in the absorption of a larger portion of the solar spectrum, as shown in figure 16(b). It is thus clear that a tradeoff exists between the maximum voltage and the maximum current density from an OPV [54]. Figure 17. Typical data obtained from a J-V measurement on an OPV cell with the dark current indicated by the black line and the AM1.5G illuminated indicated in red. The Jsc, Voc and MPP are marked as a reference. In order to generate power, the device needs to be operated in the forward bias regime. In this regime, the solar-induced current will compete with the dark current (as measured on a non-illuminated dark OPV and indicated by the black line in figure 17), which is due to charge injection in the diode structure created by the asymmetric electrodes. A typical current density – voltage (J-V) measurement performed on an AM1.5G illuminated organic solar cell is shown in figure 17 (red curve). This current density represents the net of the generated and the dark currents. The point where the generated and dark currents balance, and no net current is produced by the device, is termed the open-circuit voltage (Voc). Consequently, when the voltage measured over the device is zero, no net current is injected from the electrodes. The current density measured at this 22 point is termed the short-circuit current density (Jsc). The total generated power is the product of the current and voltage; hence no power is generated by the device at Jsc or Voc. Instead, the maximum power point (MPP) defines the point where the product between current and voltage is maximized, as visualized by the green rectangle with dashed borders in figure 17. In an ideal device, the MPP would equal the product of Jsc×Voc (red square with solid borders in figure 17) but because of, e.g., the dark current the measured MPP is reduced. The ratio between the MPP and the ideal (Jsc×Voc) is defined as the fill factor (FF). In the end, the most important metric for a PV cell is its total power conversion efficiency (PCE), as calculated by PCE J sc Voc FF Pi n (8) where Pin is the total power density of the of the light impinging upon the surface of the PV cell, which commonly is 1 kW m-2 in experiments [45]. A lot of effort has been invested on optimizing materials and processes in order to maximize the PCE of BHJ-OPV cells [55]. Record values for the PCE are however almost exclusively measured during a short time period following device fabrication, but a practical device should maintain this level of performance for an extended period of light illumination. This requirement is not simple for a BHJ-OPV cell, since the temperature increase during continuous operation can drive the phase separation beyond its optimum, and in the process reduces the device efficiency. A fabrication process aimed at locking the film morphology at its optimum is thus essential for long-term continuous operation. Several methods have been suggested with varying success and chapter 6.2 will present a fully solution-processable alternative for solving this problem. 23 4. Fullerene photochemical transformation Figure 18. The photochemical dimerization of PCBM. The fullerene C60 has been demonstrated to photochemically transform into dimers, oligomers and polymers during exposure to light [18, 56]. It has also been shown that the C60 derivative PCBM can be dimerized during exposure to green laser [57] and UV [58] light. The two fullerene monomers are connected to a dimer in a [2+2] cycloaddition dimerization reaction, as depicted in figure 18. 4.1. Raman spectroscopy Figure 19. Raman spectra showing the Ag(2) vibrational mode for PCBM films. While the pristine film (a) indicates a fully monomeric film, both the green laser exposed (b) and UV exposed (c) films shows a clear downshift, indicating the presence of PCBM dimers. 24 The presence of dimerized PCBM in a film can be established with Raman vibrational spectroscopy. In this technique, the sample is exposed to a monochromatic excitation light, which is scattered by the molecules in the sample. A small portion of the excitation light will interact with the vibrational modes (phonons) of the molecule, resulting in an inelastic scattering process where the scattered photon is shifted either up or down in energy by an amount corresponding to the phonon energy. So by measuring the scattered light spectrum it is possible to obtain information about the vibrational modes of a molecule. The Ag(2) pentagonal pinch mode gives valuable information about the degree of polymerization of fullerenes. It is located at 1469 cm-1 for monomer C60, but is down-shifted by ~5 cm-1 for each chemical connection to the C60 cage. For the C60 molecule, one downshift thus indicates the presence of dimers, two downshifts indicate linear oligomers/polymers, and three downshifts indicate a branched polymeric structure [18, 59-62]. Since a solubilizing side-group is chemically attached to the C60 cage in PCBM, the Ag(2) mode of the PCBM monomer is located at 1465 cm-1, i.e. downshifted by 4 cm-1 in comparison to the C60 monomer [57]. The Raman spectrum for a 100 nm thick pristine PCBM film is shown in figure 19(a), as obtained using a Raman excitation wavelength of λRaman = 780 nm at a power density of PRaman < 10 mW cm-2. The photochemical transformation of the PCBM film was performed by exposure to either a green laser (λ = 532 nm) or a hand-held UV lamp (λ = 365 nm), and the corresponding Raman spectra are presented in figure 19(b) and figure 19(c). Both spectra show a clear downshift of the Ag(2) mode by 5 cm-1 as highlighted by the two dotted vertical lines, which indicate the formation of PCBM dimers in both films. Interestingly, no further degree of polymerization is observed in the exposed films. The lack of a higher degree of polymerization of PCBM is motivated by the steric hindrance of the PCBM side-group [57]. 4.2. Fullerene patterning by resist-free lithography An exciting and useful property of the photochemically transformed PCBM dimers is that their solubility in organic solvents, as compared to the PCBM monomers, is drastically reduced. Thus, by selectively exposing certain regions of a pristine PCBM film, it is possible to create lateral regions of lowered solubility which can be developed by immersion into a tuned solution [57, 58, 63-65]. This process is known as photolithography. 25 Figure 20. Schematic representation of the key steps in the resist-free photolithographic patterning of PCBM: (a) film deposition, (b) light exposure through a shadow mask, (c) development in a tuned developer solution, and (d) the patterned PCBM dimer film. The optical micrographs in (e-g) display (dark brown) patterned PCBM, in the form of lines with different line widths and a square 2D structure. The resist-free photolithographic patterning of a PCBM film is schematically depicted in figures 20(a-d). The process begins with the deposition of a PCBM film through spin-coating, as shown in figure 20(a). The as-cast film is then exposed to light through a shadow-mask, featuring the pattern to be transferred as the open areas, as shown in figure 20(b). The exposure will transfer the pattern of the shadow mask into PCBM dimer regions with reduced solubility. By immersing the exposed film into a carefully tuned developer solution (e.g. chloroform:acetone in a 1:7 volume ratio), as shown in figure 20(c), the monomer PCBM is washed away, leaving only the patterned dimers on the substrate, as shown in figure 20(d). Optical micrographs of patterned (dark brown) PCBM films on SiO2 substrates are shown in figures 20(e-g). The line width of the patterned PCBM lines decreases from 100 µm to 10 µm, in steps of 10 µm, in figure 20(e), and from 10 µm to 1 µm, in steps of 1 µm, in figure 20(f). Figure 20(g) shows a 22.5 µm square pattern. The successful patterning of the smallest 1 µm sized lines in figure 20(f), and the sharp corners in figure 20(g), demonstrate the method’s capacity for high resolution. Moreover, the 26 measured line-edge roughness of 56 nm, in combination with that the smallest feature on the shadow-mask was 1 µm, indicates that an even smaller feature size of <1 µm should be achievable. 4.3. Patterning efficiency Figure 21. (a) Optical micrograph of a patterned PCBM film, with only the left part of the film being exposed. (b) An AFM height scan was performed in a direction perpendicular to the patterned interface, as indicated by the dotted line in (b). (c) The UV-vis absorption spectrum of a pristine PCBM film. (d) The normalized retained PCBM film thickness as a function of exposure dose, as extracted from AFM measurements. A mercury lamp (solid squares) and a green laser (open circles) were used for the exposure in (d). In order to evaluate the photochemical dimerization efficiency for PCBM, a set of pristine PCBM films were exposed to light in varying doses. Half of the sample was masked during the exposure, resulting in a step profile following the development step. The normalized retained thickness of the developed 27 film was measured by atomic force microscopy (AFM), as depicted in figure 21(a) and figure 21(b). Since the developer solution is assumed to selectively remove the PCBM monomers, the normalized retained thickness can be viewed as an estimate of the dimerization fraction in the exposed and developed film. Due to the wide absorption of PCBM, as shown in the UV-vis absorption spectrum in figure 21(c), a wide range of wavelengths are able to photochemically transform PCBM monomers. From a resolution perspective, it is preferable with a short wavelength since the minimum feature size in photolithography is dependent on the wavelength of the exposure light. Additionally, PCBM shows a significantly higher absorption coefficient in the UV region than in the visible region, with the anticipated consequence being that the dimerization process should be more efficient with UV-light exposure. A hand held mercury UV-light source (λ = 365 nm, P = 8.4 mW cm-2) and a longer wavelength green laser (λ = 532 nm, P = 61.3 mW cm-2) were chosen to investigate the effect of the wavelength on the process efficiency. The normalized retained PCBM film thickness as a function of exposure dose and wavelength is shown in figure 21(d), with the exposure dose being controlled by the exposure time. We quantify the process efficiency with the dose required for 50% retained thickness. It is clear that the UV exposure (solid squares) requires less energy to fully dimerize the PCBM film than the green laser (open circles), as quantified by that the 50% dose for the former process is 3.57 J cm-2 and 26.1 J cm-2 for the latter process. Some assumptions are necessary in order to transform the measured dose into a more conceptually accessible “dimerization events per absorbed photon” as represented by the symbol Φ. First, the intensity of the exposure light is uniform throughout the film thickness, and no reflections at the film or substrate surface are present, implying that the incoming light passes through the film once. Second, the absorption coefficient remains constant (equal to that of pristine PCBM) throughout the entire exposure step. Third, a successful dimerization event requires that two neighboring PCBM monomers are simultaneously excited by one photon each. Based on these assumptions it is possible to formulate an expression for Φ based on a modified version of Beer-Lambert's law: 28 N Φ di m N a bs VN A E tot M hc (9) 1 e d where Ndim is the minimum number of photons necessary to fully dimerize the PCBM film, i.e. assuming one absorbed photon per PCBM monomer. It is given by the density of PCBM (ρ = 1.5 g cm-3), the volume of the dimerized PCBM film (V), Avogadro's constant (NA) and the molar weight of PCBM (M). Nabs represents the measured number of absorbed photons for full dimerization. It is given by the total energy of the incoming light (Etot), the wavelength of the exposure light (λ), Planck's constant (h), the speed of light in vacuum (c), the absorption coefficient of PCBM at the exposure wavelength (α), and the PCBM film thickness (d). By including the relevant numbers into equation (9), notably Nabs,365 = 2.4×1019 cm-2 and Nabs,532 = 1.3×1020 cm-2, we find that the photon-todimerization efficiency is: Φ365 = 4.2×10-4 and Φ532 = 7.4×10-5. The fact that Φ365 > Φ532 highlights the advantage of UV light over visible light as it both results in a more efficient dimerization process and a higher resolution. 4.4. The photochemical reaction mechanism Figure 22. (a) Jablonski diagram showing the PCBM energy levels and the proposed reaction pathways for PCBM dimerization. (b) Normalized residual film thickness of exposed and developed PCBM films, as a function of the exposure dose and intensity. The exposure was effectuated by a high- 29 power UV-diode (λpeak = 365 nm) using different intensities and exposure times. Understanding the underlying mechanism of the PCBM photo-dimerization reaction is crucial in order to optimize the exposure conditions. The energy levels of PCBM and the possible reaction pathways during electronic excitation, relaxation and dimer formation are presented in the Jablonski diagram in figure 22(a). The energy levels are the singlet ground state (M), the excited singlet state (1M*), the excited triplet state (3M*), and the PCBM dimer state (D) [18, 66], and the transitions between the different states are marked with arrows and associated rate constants, k. The PCBM fluorescence (1M* → M) is in comparison to the intersystem crossing to the triplet state (1M* → 3M*) a very slow process. It is thus reasonable to state that k2 is negligible [18, 66]. This assumption has the consequence that all excited electrons end up in the 3M* state, and this entire transition can be described by the combined rate constant k13. C60 dimers begin to dissociate into monomers at a high temperature of 400 K [18, 62], and we therefore assume that the PCBM dimer dissociation rate k6 can be neglected during an exposure step executed at room temperature. Once a PCBM monomer is excited to the 3M* state, it can either relax to the ground state (3M* → M) with a rate of k4, or form a dimer. The latter dimerformation process is described by two alternative pathways, the uni-excited reaction and the bi-excited reaction, as described by the rate constants k5 and k’5, respectively. In the uni-excited reaction, a dimer is formed by the combination of one fullerene monomer in the triplet excited state and another neighboring fullerene monomer in the singlet ground state, i.e. 3M* + M → D. Whereas in the bi-excited reaction, two neighboring monomers need to be simultaneously excited in order to form a dimer: 3M* + 3M* → D. A Raman spectroscopy study by Eklund et al. came to the conclusion that the uni-excited reaction described the dimerization rate of C60 fullerenes [18]. It is however here contradicted by the data in figure 22(b), which shows that higher-intensity exposure light results in a lower exposure dose for the dimerization of a PCBM film. The uni-excited reaction pathway, which only requires that one monomer is excited in order to form a dimer, will show an intensity-independent dose, and it can therefore not be the accurate description of the behavior seen in figure 22(b). The bi-excited reaction pathway, which requires two neighboring monomers to be simultaneously excited, will feature the observed intensity-dependence of the dose. It is therefore concluded that the bi-excited reaction pathway describes the photochemical dimerization process of PCBM. 30 The detailed understanding of the reaction processes for PCBM dimerization results in the practical conclusion that high-intensity UV-light is an ideal light source for the exposure step. The short wavelength will ensure high resolution, strong absorption and low dose. For example, short wavelength UV-light, with a wavelength of 365 nm and an intensity of 8000 mW cm-2, will result in an exposure time of 1 mere second. This short exposure time fits well with large-scale processes, such as a roll-to-roll (R2R) fabrication. 4.5. PCBM OFETs Figure 23. Transfer (left) and output (right) data for typical PCBM OFETs, as measured at different instants during the patterning process. The transfer data were measured at VDS = 60 V. The output data were measured in VGS steps of 20 V, starting from 0 V (lower trace) and ending at 80 V (upper trace). 31 The impact of UV-patterning on the electronic transport properties of PCBM was investigated with BG-TC OFETs, comprising a PCBM film as the active material, Au as the source and drain electrodes, and SiO2/p:Si as the gateoxide/gate; the experimental details are presented in Paper I. The transfer and output characteristics are presented in the left and right part of figure 23, respectively, as measured on: (i) pristine PCBM, (ii) UV-exposed PCBM (λ = 365 nm, P = 8.4 mW cm-2, t = 15 min), (iii) UV-exposed and developed PCBM, and (iv) unexposed and developed PCBM. Treatment (cm2 µn V-1 s-1) [a] VT (V) Ion/Ioff (a.u.) Pristine 1.8(±0.6)×10-2 2 2.0×104 Exposed 1.6(±0.5)×10-2 7 1.2×104 Exposed + Developed 1.2(±0.2)×10-2 10 1.2×104 --- --- --- Solely Developed [a] Mean value with standard deviation in parenthesis Table 1. Performance metrics of PCBM transistors as measured during the UV-patterning procedure. The transistor data demonstrate well-behaved n-type OFETs for (i)-(iii), with monotonously increasing IDS with increasing VGS in the transfer characteristics, and clear saturation in the output characteristics. As expected, the solely developed sample shows no OFET characteristics, simply because the unexposed PCBM were dissolved and removed by the developer solution. Both µn and VT were extracted from the transfer data, as described in section 3.1, and the Ion/Ioff ratio was calculated as the ratio of IDS at (VDS = VGS = 60 V) and (VDS = 60 V, VGS = 0 V). The average values from >60 characterized OFETs are summarized in table 1. To our satisfaction, PCBM features a respectable mobility following patterning, although it is slightly reduced in comparison to the pristine state. We tentatively attribute the decrease to a reduction of intermolecular π-orbital overlap following the transformation of some sp2 orbitals to sp3 orbitals; see figure 18. Interestingly, the dimerization also makes PCBM less sensitive to ambient air. By exposing the OFETs to ambient air for 24 h prior to the FET characterization, we found that the mobility of pristine PCBM dropped to a mere 1 % of its initial value, whereas the UV-patterned PCBM retains 39 % of its initial mobility. We speculate that this improvement is due to a more 32 dense and possibly more crystalline film, which could act as an improved barrier towards O2 permeation into the active layer [58, 67]. Most importantly, the electronic mobility of PCBM (and C60) is only slightly reduced by the photochemical transformation [57, 63]. This fact indicates that high-resolution and resist-free patterning of the fullerene-based active material in organic electronic devices could be possible. In OFETs, this advantage could be used to reduce inter-device and fringe-field leakage currents, and to realize advanced circuits such as memory devices. 4.6. Realizing complementary logic circuits with resist-free patterning The resist-free patterning technique can also be utilized to realize complementary circuits with a single-layer active material, comprising a blend of n-type PCBM and p-type poly(3-hexylthiophene-2,5-diyl) (P3HT). By selecting an active material with high PCBM content, a predominantly ntype device is obtained. Interestingly, it is possible to transform this device into featuring p-type behavior, by immersing the device into a developer solution tuned to selectively dissolve (non-exposed) PCBM monomers, but not (exposed) PCBM dimers and P3HT. By using this single-layer blend film as the common active material for an array of transistors, exposing some of the transistors to high-intensity light, and then developing the entire array an interesting and attractive result is obtained. The exposed and developed transistors remain n-type, as the active material is left effectively intact by the developer solution, while the solely developed transistors have transformed into being p-type, following the removal of the (non-exposed) PCBM monomers [64]. 33 4.6.1. The inverter Figure 24. (a) Photograph and (b) schematic diagram of the PCBM:P3HT complementary inverter fabricated with the resist-free patterning technique. The output data of (c) the p-type FET and (d) the n-type FET, with VGS changed in steps of 10 V starting from 0 V and ending at 50 V and VDS swept at a rate of 2 V s-1, as indicated by the arrows. (e) The static and (f) the dynamic inverter characteristics, as recorded with VDD = 50 V and VDD = 44 V, respectively. 34 A schematic of the complementary inverter circuit, comprising a p-type and an n-type FET, is depicted in figure 24(b). The FETs were fabricated in a BGTC geometry, comprising a common active material of PCBM:P3HT in a mass ratio = 5:1, Au as the source and drain electrodes, and SiO2/p:Si as the gate-oxide/gate. The active material in the upper and lower FETs was then tuned to be p-type and n-type, respectively, following the procedure outlined in the previous section. More specifically, a green laser (λ = 532 nm, P = 60 mW cm-2, t = 30 min) selectively exposed the active material in the FET designed to be n-type, where after the entire active material film was developed in the developer solution. Figure 24(a) shows a photograph of two inverters, with the exposed dimer-PCBM:P3HT and the unexposed P3HT regions distinguished by their dark blue-green and red-brown appearances, respectively. A typical output device performance of the p-type and n-type OFETs are shown in figure 24(c) and figure 24(d), respectively. Both devices show wellbehaved operation with clear current saturation and low hysteresis. A slight ambipolarity of the n-type FET is observed as a slight increase in IDS at high VDS as VGS approaches 0 V. The µ and VT extracted from the transfer measurements on 5 pair of transistors are shown in table 2. Device µ (cm2 V-1 s-1) [a] VT (V) [a] p-FETPUN 1.3(±0.4)×10-4 -14.8(±2.5) a-FETPDN 1.7(±0.8)×10-4 19.4(±1.7) [a] Mean value with standard deviation in parenthesis Table 2. Performance metrics of the two OFETs forming the inverter. Figure 24(e) shows the static inverter characteristics, as recorded at VDD = 50 V. The inverter features a sharp switch of Vout (blue solid line) at Vin ≈ 30 V, as quantified by a high gain of g = 11 ±1. The IDD (red dotted line) shows a peak at the switching threshold, as expected, a low leakage current of <1 nA at Vin > 35 V, and a larger leakage current for Vin < 25 V. The latter is an effect of the slightly ambipolar character of the n-type FET, which results in a direct current path from VDD to GND. Figure 24(f) shows the dynamic inverter behavior, as measured at VDD = 44 V. The high-to-low transition (tpHL = 2.9 s) is delayed by an overshoot of Vout, as it is pulled towards GND by the n-type FET. The low-to-high transition (tpLH = 0.6 s), in contrast, shows an ideal behavior as Vout is pulled towards 35 VDD by the p-type FET. A total propagation delay of 1.75 s is measured at a VDD of 44 V. 4.6.2. The ring oscillator Figure 25. (a) Schematic of a 5-stage ring oscillator realized with the complementary PCBM:P3HT inverters, and (b) the corresponding temporal behavior of Vout and IDD at VDD = 50 V. (c) the delay time and the average power consumption as a function of supply voltage, and (d) the PDP and EDP as a function of the supply voltage. A 5-stage ring oscillator was fabricated by connecting 5 inverters in series, with the output of the last inverter connected to input of the first, as depicted schematically in figure 25(a). When the supply voltage was connected, the circuit began to oscillate, and temporal data on Vout and IDD at VDD = 50 V are shown in figure 25(b). The frequency of operation is 10 mHz, and the circuit consumes an average IDD ≈ 15 nA. Interestingly, the measured Vout only oscillates between 25-50 V and thus never operates in the ambipolar region. 36 Figure 25(c) shows that the delay time, tp, decreases and the average power consumption, Pav, increases with increasing VDD, as expected, for the 5-stage ring oscillator. The superlinear increase of Pav with increasing VDD indicates that the power consumption can become an issue at high VDD. The dependence of the power delay product, PDP, and energy delay product, EDP, on VDD are shown in figure 25(d). A PDP minimum of 2.0 µJ is found at VDD = 46 V, indicating that this is the most efficient VDD for operation. However, no minimum is found for EDP in the measured interval, since the reduction of tp is greater than the increase of Pav as VDD increases. This indicates that the supply voltage for optimum operation will be found at VDD > 50 V. In summary, it has been shown that functional complementary inverters and ring oscillators can be fabricated with a facile photolithographic procedure developed in-house, without the use of a sacrificial photo-resist material. By considering the high values for the intrinsic capacitance (Ci = 14.7 pF) and the extrinsic capacitance (CL = 1.44 ±0.15 nF) for the inverters in this particular circuit, it is clear that further improvements in terms of speed and power consumption by several orders of magnitude can be achieved by reducing these parasitic capacitances. 37 5. Fullerene C60 nanorods For facile electron transport, a crystalline material is often preferred over its amorphous counterpart [14-16, 68]. Self-assembly is a common method for the fabrication of crystalline organic materials with controlled size and shape, and crystalline C60 structures ranging from 0-D dots, through 1-D rods, to 2-D discs have been realized using such methods [17, 69-73]. A notably straightforward solution-based self-assembly fabrication method is the liquid-liquid interfacial precipitation (LLIP) method, which allows synthesis of crystalline 1-D C60 nanorods (NRs) with sub-µm diameter [7480]. 5.1. Synthesis The LLIP synthesis of C60 NRs is a 3-step process: i) interface formation, ii) nucleation, and iii) crystal growth. An example of a fabrication process is detailed below: C60 is dissolved in a good solvent (1 mg ml-1 in mdichlorobenzene, m-DCB) and 1 ml of the solution is added to a glass vial. 10 ml of a poor C60 solvent (ethanol) is gently added to the vial, so that an interface is formed between the good and poor solvents; see left vial in figure 26(a). The two-component solution is ultrasonicated to induce partial mixing of the two solvents at the interface. The mixing results in local oversaturation of C60, so that nucleation and precipitation of C60 seed particles can take place, as manifested in the greenish interfacial region in the middle vial in figure 26(a). The hermetically closed vial is then left at room temperature, and after 2 days C60 NRs are observed to begin to deposit at the bottom of the vial; see right vial in figure 26(a). After 3 days the crystal growth is terminated by vigorously shaking the vial and the resulting asgrown NRs are ready for use. An optical micrograph and a transmission electron microscope (TEM) image of a batch of as-grown NRs are shown in figure 26(b) and figure 26(c), respectively. The NRs feature a hexagonal cross section, lengths of ~1 mm, an aspect ratio of >103, and they are highly flexible [81]. The size of the NRs is controlled by the number of crystal seeds formed during the nucleation step. This number can be controlled by the solvent intermixing during the ultrasonication, and an increased ultrasonication power results in enlarged intermixing and an increased number of crystal seeds, which in turn results in smaller NRs [82]. We have utilized two different ultrasonication powers for the fabrication of two types of NRs with: (i) a mean diameter of Ø1 = 250 nm, within a range of 100-400 nm (Paper III); (ii) Ø2 = 172 nm, within a 38 range of 50-360 nm (Paper IV). The size distribution of the latter is presented in figure 26(d). Figure 26. (a) Photograph of vials comprising solutions in the different stages of the LLIP process: (left) interface formation, (middle) nucleation, and (right) crystal growth. (b) Optical micrograph and (c) TEM image of the as-grown NRs. (d) Diameter distribution of NRs with an average diameter (Ø2) of 172 nm. 39 5.1.1. Polymerization Figure 27. Schematic representation of the photochemical transformation of NRs into PNRs. The photograph in the inset shows vials containing dispersions of as-grown NRs (left) and PNRs (right), depicting the color shift from green-brown to red-brown following the exposure. Exposing the as-grown NRs to high-intensity light transforms the monomeric C60 in the as-grown NRs into polymeric C60. The exposure was performed by directing a laser beam (λ = 532 nm, P = 15 mW mm-2, t = 20 h) into the NR dispersion through the bottom of the vial, as depicted in figure 27. The width of the laser beam was adjusted to match the width of the vial (~1 cm), and the sides of the vial were covered in Al foil to trap the light inside the vial. The exposed NRs featured a color shift from green-brown to red-brown, as seen in the photograph inset in figure 27, while the NR diameter distribution was found to be unaffected. The photochemically transformed NRs will hereafter be referred to as polymerized nanorods (PNRs). It should be noted that the exposure was performed on NRs in dispersion, making it a scalable process. 40 5.2. Characterization The NR structure was characterized by TEM, high-resolution TEM (HRTEM), scanning TEM (STEM), Raman spectroscopy, x-ray diffraction (XRD), and selective area electron diffraction (SAED), while the thermal characterization was performed using thermal gravimetric analysis (TGA). 5.2.1. As-grown Figure 28. Characterization of as-grown NRs with (a) XRD and (b) HRTEM, with the (top left) inset showing a SAED image. Figure 28(a) shows the XRD of as-grown NRs, with the peak pattern indicating a hexagonal close-packed (hcp) structure, with a unit cell size of a = b = 23.71 Å and c = 10.18 Å. The sharp distinct peaks indicate a singlecrystal structure, which is supported by both the HRTEM image in figure 28(b), in which both crystalline faults and dislocations are absent, and the SAED pattern in the inset in figure 28(b). It is thus concluded that the asgrown NRs feature a single-crystalline hcp crystal structure. TGA of as-grown NRs reveals a linear weight loss of 10.7 % between 85-171 °C, as shown in figure 29(a). This weight loss is assigned to the evaporation of guest m-DCB molecules from within the NRs. The weight loss with an onset of ~400 °C is due to the sublimation of C 60, which features a sublimation temperature of 390 °C in the solid state. The Raman spectra of as-grown NRs and pristine C60 are displayed in the upper red curve and lower blue curve in figure 29(b), respectively. The asgrown spectrum features 10 characteristic C60 peaks (8 Hg and 2 Ag) and 3 additional peaks, as marked with asterisks, and these additional peaks can be 41 removed by annealing at 200 °C. Furthermore, the Hg(1) and Ag(1) modes of the as-grown NRs are downshifted in comparison to pristine C60 (see inset in figure 29(b)). The new and shifted Raman features of as-grown NRs are assigned to the existence of solvated m-DCB guest molecules in the NR lattice [72, 83, 84]. Figure 29. (a) TGA of as-grown NRs. (b) Raman spectra of as-grown NRs (i, red upper curve) and pristine C60 (ii, blue lower curve). The inset in (b) highlights the downshift of the Hg(1) and Ag(1) modes for as-grown NRs (i) in comparison to pristine C60 (ii). 42 5.2.2. Annealing Figure 30. (a) XRD patterns of annealed NRs (i, red upper curve) and pristine C60 (ii, blue lower curve). (b) STEM, (c) HRTEM, and (d) SAED of the annealed NRs, which reveal the existence of a porous and polycrystalline structure. A vacuum annealing step (T = 200 °C, p <1 kPa, t = 24 h) was introduced in order to remove the solvated m-DCB molecules, and the upper red curve in Figure 30(a) shows the XRD of annealed NRs. Interestingly, the crystal structure has transformed from hcp to face-centered cubic (fcc), with a unit cell size of a = b = c = 14.13 Å; i.e. the same crystal structure as pristine C60 (blue lower curve) [70]. Further inspection of the annealed NRs by STEM (figure 30(b)), HRTEM (figure 30(c)) and SAED (figure 30(d)) reveals the existence of a porous polycrystalline structure, with clearly distinguishable crystal grain boundaries. 43 5.2.3. Polymerization Figure 31. The Ag(2) Raman mode of PNRs (upper panels, solid line) and pristine C60 (lower panels). The deconvoluted peaks of the PNRs (dotted lines) are indicated in the upper panels. The excitation laser wavelength was 514 nm in (a) and 785 nm in (b), with the longer wavelength probing deeper into the sample. Figure 31 shows the Ag(2) Raman mode of PNRs (upper panels, solid line) and pristine C60 (lower panels), with the deconvoluted PNR peaks indicated by dotted lines in the upper panels. The excitation wavelength was λRaman = 514 nm and λRaman = 785 nm in figure 31(a) and figure 31(b), respectively. Since the C60 absorption is significantly higher at 514 nm than 785 nm, the former will probe the surface and the latter the bulk. A comparison of the deconvoluted peaks of the Ag(2) mode shows a relative larger downshift at 514 nm, which is assigned to a higher degree of polymerization on the surface than in the bulk. In other words, the PNRs can be described as comprising a highly polymerized shell surrounding a mainly monomeric core, as schematically indicated in the cartoon above the figure. In order to investigate the effects of polymerization on the solubility, both as-grown and PNRs were filtered out of the synthesis dispersion and added to pure m-DCB in a concentration of 1 mg ml-1. The as-grown NRs were observed to readily dissolve in the good C60 solvent, as evident by the formation of a characteristic clear purple solution; see left vial in the photograph in figure 32(a). Interestingly, the PNRs remained intact and well dispersed in the m-DCB, as evident by the opaque brown dispersion in the 44 right vial in figure 32(a). In fact, subsequently recorded TEM images of the PNRs reveal that these feature a completely smooth surface and are completely unharmed by the good C60 solvent (see figure 32(b)). Similar tests using common hydrophobic solvents showed similar results, indicating that the PNRs can be processed in a wide range of organic solvents without damage. Figure 32. (a) Vials containing dissolved as-grown NRs (left vial with purple solution) and dispersed PNRs (right vial with brown dispersion) at a concentration of 1 mg ml-1 in m-DCB. (b) TEM image of PNRs, with the inset showing a single PNR at higher magnification. 45 5.3. The C60 nanorod transistor Figure 33. Schematic showing the NR FET structure, with the NRs aligned in the shortest sourced-drain direction, i.e. perpendicular to the sourcedrain interfaces. OFETs with NRs as the active material were realized in order to evaluate the electronic transport properties of the NRs in the pristine state and following the structural changes induced by annealing and photo-polymerization. A schematic of the employed BG-TC device structure is shown in figure 33, with p:Si/SiO2 (200 nm) as the gate/gate-oxide, and Cr/Au (15/15 nm) as the source-drain electrodes. In order to accurately measure µn with equation (3), it is important to accurately estimate the effective WD and LG of the NRs involved in the transistor operation i.e. the NRs bridging the gated channel between the source and drain electrodes. The WD can be readily calculated as the product of the mean NR diameter (Ø) and the observed number of NRs crossing the channel, while the estimation of LG benefits from the alignment of the NRs in the shortest source-drain direction; see figure 33. This geometry will also produce the largest source-drain current. 46 5.3.1. Nanorod deposition Figure 34. Schematic representation of the drop-casting of as-grown NRs from an ethanol/m-DCB dispersion onto a transistor structure, highlighting (a) the initial ethanol evaporation and (b) the subsequent mDCB evaporation and the formation of a random network of NRs and needle-like C60 crystals. The NRs can conveniently be deposited by drop-casting, whereby a (~25 µl) drop of NR dispersion was applied onto the transistor structure. The dispersion comprises a solvent mixture of m-DCB and ethanol, with the two solvents differing greatly in vapor pressure (0.252 kPa versus 7.87 kPa at 25 °C) [85]. As a result, the high-vapor pressure ethanol will evaporate much faster during drying, leaving the slow-drying m-DCB on the substrate, as depicted in figure 34. As a consequence, some of the NRs will dissolve in the remaining m-DCB solvent, while the remaining undissolved NRs will deposit randomly on the substrate. The dissolved C60 is observed to exclusively deposit on the Au electrodes in the form of needle-like structures, presumably due to the large surface energy difference between the bare SiO2 channel and the Au electrodes. The random positioning of the NRs have the unfortunate consequence that the effective LG of drop-cast deposited NRs is difficult to determine with good accuracy. To circumvent these issues, the method of controlled dip-coating was utilized. The transistor substrate was immersed into the NR dispersion with the long side of the transistor channel parallel to the solution interface, and then slowly withdrawn at a fixed angle of 70° and a constant rate of 3 cm min-1; see schematic in figure 35(a). A photograph of an aligned NR in the transistor channel is shown in the photograph in figure 36(a). The corresponding angular distribution of the dip-coated NRs is shown in figure 35(b), where the measured distribution of the drop-cast NRs is included for comparison as the dashed blue line. It is clear that dip-coating results in a 47 good alignment of the NRs in the transistor channel and that the issue of C 60 needle formation is effectively resolved. It is thus reasonable to approximate LG with the shortest source/drain electrode distance in the calculation of the mobility in transistor measurements on dip-coated NRs. Figure 35. (a) Schematic of the dip-coating procedure and an optical micrograph of dip-coated NR on a transistor structure (the light and dark regions correspond to the Au electrodes and the SiO 2 channel, respectively). (b) Angular distributions of the dip-coated NRs and drop-cast NRs, relative to the channel. 48 5.3.2. Transistor performance Figure 36(a) presents typical transfer characteristics of as-grown NRs (Ø1 = 250 nm) deposited by drop-casting, before and after annealing. The undesired large hysteresis is suppressed after the annealing, and this improvement in performance is assigned to the removal of the chargetrapping solvent molecules from the NRs. We have chosen to not include data on the absolute mobility of the drop-cast NRs, due to the difficulty in establishing the correct value for the gate length, LG, in drop-cast NR-based OFETs, as discussed in detail in section 5.3.1. Figure 36. Transfer characteristics of (a) drop-cast and (b) dip-coated NR OFETs in different states. (c) Mobility distribution of as-grown NRs and PNRs. (d) Output characteristics of a dip-coated PNR OFET. Figure 36(b) shows the transfer characteristics of OFETs based on dipcoated NRs (Ø2 = 172 nm) as the active material, with the NRs either being characterized as-grown or after photo-induced polymerization. Quantitative performance data for dip-coated NRs as a function of nanorod diameter, annealing and photo-polymerizaton are summarized in table 3. We find that the highest mobility is recorded on as-grown NRs with the lower average diameter of 172 nm. Both the annealing and photo-polymerization 49 procedures result in a significant drop for both the mobility and the on/off ratio. We assign this reduction to the introduction of grain boundaries during annealing and to the formation of lower-mobility C60 polymers during photo-polymerization. Treatment Ø (nm) µn (cm2 V-1 s-1) [a] VT (V) [a] Ion/Ioff (a.u.) As-grown 250 4.4(±2.7)×10-2 17.4(±3.5) 103 Annealed 250 8.8(±2.6)×10-3 18.9(±1.7) 102 As-grown 172 3.0(±2.9)×10-1 21.5(±3.8) 104 Polymerized 172 4.7(±3.9)×10-3 20.4(±5.5) 103 [a] Mean value with standard deviation in parenthesis Table 3. Transistor data on dip-coated NR OFETs. We also point out that the data in table 3 are average values (and standard deviations), and that the variation in performance can be rather significant, as demonstrated by the mobility distributions for as-grown and polymerized NRs in figure 36(c). In fact, the best-performing NR-OFET, comprising asgrown NRs with an average diameter of 172 nm, did break the mobility "unity" barrier at 1.0 cm2 V-1 s-1. This proves that solution-processable NRs can compete with state-of-the-art vacuum-deposited C60 and a-Si [6, 86-88]. We further note that the drop in mobility following photo-polymerization is in agreement with previous studies on the effects of chemical transformation of C60 [63, 79]. In this context, it is, however, relevant to remind ourselves that the charge accumulation and charge transport in an OFET take place in the first few monolayers of the OSC deposited on the gate insulator, which for PNRs correspond to the highly polymerized shell [19, 28, 89]. It is thus reasonable to assume that the highly monomeric PNR core could exhibit a higher mobility, similar to what was measured in as-grown NRs. Typical output characteristics of a dip-coated PNR OFETs are shown in figure 36(d). The devices exhibits a good IDS saturation at high VDS, but the exponential increase at low VDS indicates a Schottky-type injection barrier at the source electrode interface [90]. This observation, in combination with the utilized simplification that all NRs are perfectly aligned in the transistor channel, implies that the documented values for the mobility are underestimating the true value [91, 92]. 50 In summary, we have shown that single-crystal NRs can be synthesized in solution using the LLIP technique, and that such NRs can be applied as the active material in OFETs with a high electron mobility. A light-exposure step can transform the NR surface to become polymeric, so that the resulting PNRs can form a stabile dispersion in common hydrophobic solvents. With these properties demonstrated, the NRs can be exploited in several applications, and we mention the decoration of PNRs with Pd catalyst particles, which resulted in an improvement in the oxidation of ethanol (Paper V). 51 6. Material design for improved photovoltaics The efficiency and practical usefulness of BHJ-OPVs are influenced by a wide range of factors, and dedicated work in the field has resulted in the demonstration of small-area devices with a PCE >10 % [55, 93, 94]. These improvements are stemming from a reduction of the Eg of the constituent compounds, so that the active material can absorb more of the incoming solar radiation, and an improved active-material phase morphology as manifested in an increased Jsc [55, 95-98]. It is also possible to attain a high output voltage from BHJ-OPVs, but such reports are comparatively sparse [55, 99]. Moreover, a well-documented issue with current BHJ-OPVs is the poor morphological stability of the active material [52, 53, 100], and the development of functional methods for the resolution of this setback is highly needed. 6.1. A small-molecule donor compound for OPVs Figure 37. (a) Molecular structure of ZOPTAN-TPA. (b) UV-vis absorption spectra of ZOPTAN-TPA in solution (open circles) and as a thin film (solid squares). The BHJ-OPV commonly comprises an active-material blend of conjugated polymer as the donor and a small-molecule fullerene as the acceptor. An important advantage with small molecules is that they are comparatively easy to synthesize and purify, and it is therefore of interest to employ smallmolecules also for the donor compound. For the attainment of a small molecule donor compound with appropriate energy levels for high OPV performance, we choose to synthesize a compound with an internal donoracceptor-donor (D-A-D) structure. By chemically connecting a (2Z,2Z)-2,2´(2,5-bis(octyloxy)-1,4-phenylene)bis(3-(5-bromothiophen-2-yl)acrylonitrile) 52 acceptor unit with two peripheral 4-(diphenylamino)phenylboronic acid donor units, we attained the small-molecule donor compound ZOPTANTPA, with its chemical structure presented in figure 37(a); for a description of the synthesis procedure, see Paper VI. ZOPTAN-TPA was thermally characterized with thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). The TGA showed that the material is stable (under inert atmosphere) up to 410 °C, whereas the DSC revealed that ZOPTAN-TPA can crystallize and that its melting point is 145 °C. Figure 37(b) shows the UV-vis absorption spectra of ZOPTAN-TPA in solution (open circles) and as a thin film (solid squares). The film features a red-shifted absorption onset in comparison to the solution, which is positioned at ~600 nm; the latter corresponds to an optical energy gap of Eg,OPT = 2.1 eV. The HOMO and LUMO levels of ZOPTAN-TPA were established with cyclic voltammetry (CV), and these data and the electrochemical energy gap (Eg,EC) and Eg,OPT are presented in table 4. Compound ZOPTAN-TPA HOMO (eV) LUMO (eV) Eg,EC (eV) Eg,OPT (eV) –5.2 –3.0 2.2 2.1 Table 4. Measured energy levels and energy gaps for ZOPTAN-TPA. By comparing the LUMO level of ZOPTAN-TPA with the LUMO level of PCBM (-3.7 eV), we find that ΔELUMO = 0.7 eV in a PCBM:ZOPTAN-TPA blend. Thus, it is expected that excitons, generated in the ZOPTAN-TPA phase, will be efficiently split up at the ZOPTAN-TPA/PCBM interfaces in a BHJ-OPV device. Moreover, as the difference in energy between the LUMO of the PCBM acceptor and the HOMO of the ZOPTAN-TPA donor is notably large at 1.5 eV, and this value dictates the maximum for the open-circuit voltage, Voc, (see section 3.2.3), we also anticipate the opportunity for a high output voltage. BHJ-OPV cells were fabricated with an ITO/PEDOT:PSS anode and an Al cathode sandwiching a PCBM:ZOPTAN-TPA active layer (see Paper VI for the experimental procedure). The average photovoltaic performance of these 7×3 mm2 devices as a function of active material composition is presented in figure 38(a)-(e). As anticipated, a high Voc was attained, with the maximum of 1.03 V obtained for a PCBM:ZOPTAN-TPA mass ratio of 1:1. The Jsc, and accordingly the EQE, featured a peak in performance for the 2:1 mass ratio. The FF is relatively modest, with a maximum of 32 % for the 1:1 mass ratio. In order to investigate whether recombination at the cathode could be the 53 culprit to the low FF, we also fabricated and tested devices with a 2nm LiF electron-transport layer inserted between the cathode and the active materials. As this procedure did not result in a significant improvement in performance, we exclude cathodic recombination as the culprit. With the information of Voc, Jsc and FF at hand, it is straightforward to calculate the PCE, which features a peak value of 1.17 % for the 2:1 mass ratio. Figure 38. (a) The open-circuit voltage, (b) the short-circuit current density, (c) the fill factor, (d) the power conversion efficiency, and (e) the external quantum efficiency for ITO/PEDOT:PSS/PCBM:ZOPTAN-TPA/Al solar cells as a function of the PCBM:ZOPTAN-TPA mass ratio in the active material. We also wish to emphasize that the data in figure 38 represented the average values (and standard deviations) derived from characterization of 14 independent devices, and that it is common that the performance of BHJOPV can vary significantly between different devices. We have therefore chosen to include results from our “champion” device, being a non-annealed ITO/PEDOT-PSS/PCBM:ZOPTAN-TPA/Al OPV cell, with a PCBM:ZOPTAN-TPA mass ratio of 2:1. The champion device featured the following performance metrics: Jsc = 6.01 mA cm-2, Voc = 0.98 V, FF = 32 %, and PCE = 1.86 %. The OPV performance as a function of annealing is presented in figure 39(a). The 2:1 mass ratio was selected for this study, since it featured the best performance (see figure 38). While only minor changes in Voc and FF were found (data not shown), the Jsc, and consequently the PCE, showed major drops in performance following annealing. For instance, a 60 s annealing at 70 °C under inert N2 atmosphere resulted in a 15 % and 19 % drop in Jsc and PCE, respectively, and an increase in temperature to 90 °C resulted in a drop of 26 % and 33 %, respectively. 54 Figure 39. Relative change of (a) Jsc and (b) PCE as measured on PCBM:ZOPTAN-TPA (2:1) devices following annealing at 70 °C (solid line) and 90 °C (dotted line) for varying time. AFM micrographs of similar (c) as-prepared and (d) annealed (90 °C for 5 min) films. AFM micrographs of the same type of PCBM:ZOPTAN-TPA blend film directly after spin-coating and after a 5 min thermal annealing at 90 °C under N2 atmosphere are displayed in figure 39(c) and figure 39(d), respectively. The as-prepared film comprises small depressed features of unknown origin, with a typical vertical size of <20 nm, on an otherwise smooth film surface. With thermal annealing, the depressed features vanish and are replaced by protruding features. The size and density of these protruding features are observed to increase with both temperature and annealing time. Based on the observation that ZOPTAN-TPA can crystallize (with a melting temperature of Tm = 145 C, as established with DSC), we attribute the observed growth of the protruding features to the enrichment and subsequent crystallization of ZOPTAN-TPA within the blend film. As the lateral size of these features of ~100 nm is much larger than the typical exciton diffusion of ~10 nm in organic semiconductors, it is straightforward to assign the observed decrease of Jsc with thermal annealing to a decrease in the area of the critical charge-generating donor/acceptor interfaces in the active-material film when (too) large ZOPTAN-TPA crystallites form. 55 6.2. The C60 nanorod OPV Figure 40. Schematic device structure of an OPV based on C 60 polymerizednanorods. One severe stability issue with BHJ-OPVs originates in the propensity of the active material constituents to phase separate over time, with a reduction of the exciton dissociation efficiency, and a lowering of the Jsc and PCE being a common consequence [101, 102]. An example of this detrimental phaseseparation process was presented in the last paragraph in the previous section, where an elevated temperature, as often encountered during OPV operation, was shown to drive the undesired phase-separation process. Here, we propose that the issue with an unstable phase morphology in BHJ-OPVs could be alleviated by employing the polymerized nanorods as the acceptor material for the realization of the device schematically depicted in figure 40. We anticipate that not only will the charge-separation process be stabilized in such a device, but also the subsequent charge-transport processes be improved if the nanorods can be aligned with their long axes in the interelectrode direction. In order to investigate the feasibility of the outlined concept, we have fabricated OPVs with a blend of PNR:P3HT. The as-grown NRs were first ultrasonicated for 1 h to decrease their size to a length of 1-3 µm and a diameter of 170 nm, thereafter photo-polymerized, and finally dispersed in a P3HT-in-m-DCB solution, at a PNR:P3HT mass ratio of 1:2. This blend solution/dispersion was spin-coated onto a PEDOT:PSS/ITO/glass 56 substrate, whereafter an Al cathode was thermally evaporated on top. The performance of the OPV device was modest, but it is straightforward to realize that a significant improvement should be attainable via a decrease in NR size and through an alignment of the NRs, so that exciton split-up and charge transport can be improved. In this context, it is interesting to note that NRs with a small diameter of 50 nm can be synthesized; see figure 26(d). 57 7. Conclusions We demonstrate that the fullerene PCBM can be photochemically transformed into a dimeric state in a bi-excited reaction process, and that the exposed material features a significantly reduced solubility in common solvents as well as an effectively retained electron mobility. This attractive combination of material properties allows for a direct and resist-free lithographic patterning of electronic PCBM films down to a smallest feature size of 1 µm, using a simple and scalable two-step process constituting light exposure and solution development. In a further development, it was shown that the two-step method was useful also in the area-selective transformation of fullerene/conjugated-polymer blend films, as demonstrated through the realization of a functional complementary logic circuit in the form of a 5stage ring oscillator. In another project, we have synthesized highly flexible, single-crystal C60 nanorods with a solution-based self-assembly process termed liquid-liquid interfacial precipitation. The 1-dimensional nanorods can be deposited from their synthesis solution and employed as the active material in field-effect transistor devices. Here, it was revealed that the as-fabricated nanorods can feature an impressive electron mobility of 1.0 cm2 V-1 s-1, which is on par with the performance of a work horse in the transistor field, viz. vacuumdeposited amorphous Si. We further demonstrated that the processability of the nanorods can be improved by a tuned light-exposure treatment, during which the nanorod shell is polymerized while the high-mobility interior bulk is left intact. This has the desired consequence that stabile nanorod dispersions can be prepared in a wide range of solvents, and we anticipate that functional electronic devices, including organic solar cells, based on solution-processable nanorods can be realized in a near future. 58 Acknowledgements First and foremost I would like to thank my brilliant supervisor Ludvig Edman for giving me the chance to come to Umeå and be a part of the Organic Photonics and Electronics Group. I have truly learned a lot during the last 5 years and much of it is thanks to you. Next up I would like to thank the incredible people in the OPEG group, starting with the office squad Amir Asadpoordarvish and Andreas Sandström; may the science you are throwing at the wall be vitorious. Nikolai Kaihovirta, good luck in Turku and may your frolf discs fly far and true. A special gratitude is aimed towards my permanent collaborators Jia Wang and Jenny Enevold for countless discussions about solar cells, fullerenes, and other things. Special thanks also to the ink-maestro Shi Tang, Mattias Lindh, Petter Lundberg and Ning Sun. I would also like to acknowledge the previous group members Nahid Morshed, Jimmy Granström, Antoni Munar and Piotr Matyba. Our lab-technician Roushdey Sahl also deserves a big thanks for making all of our lives easier by keeping the labs running. I also owe gratitude towards my collaborators outside of the group, with special mentions of the brilliant Hamid Barzegar and his equally brilliant supervisor Thomas Wågberg. It was a true pleasure to work together with you. I am also grateful for the science done and discussed together with Javed Iqbal, Bertil Eliasson, Florian Nitze, Guangzhi Hu, Xueen Jia, Tiva Sharifi, Srikanth Revoju, Eduardo Gracia, Nathaniel Robinson, Deyu Tu and Robert Forchheimer. I would also like to specially thank Patric Stafshede for offering me the opportunity to join the LunaLEC crew. Together we will do great things. I would also like to thank the people I got into contact with during my teaching endeavors: Jens Zamanian, Leif Hassmyr, Hans Forsman, Gabriella Allansson, Sune Pettersson, Ludvig Lizana, Lars-Erik Svensson, Patrik Norqvist, Jonas Larsson, Isak Silander, Mats Forsberg and Michael Bradley. A big thanks also to all students. A big thanks to Jörgen Eriksson, Katarina Hassler, Lilian Andersson, Lena Burström, Karin Rinnefeldt and Nils Blix for all the help with my various silly questions and problems. Special thanks to the "bluecoats" of the workshop Lena Åström, Tomas Gustafsson and Peter Wikström for very professional help with complex demands. I would also like to acknowledge 59 the remaining staff at the department of physics for making it a great place to work. I am also grateful for the support from my friends from the south: Anil Dey (although I suppose it is a bit more west than south now), Fredrik and Veddy Hägerström and Tim Larsson. I moved 1000 km north but still you did not get rid of me. I would also like to thank all my relatives for their encouragements. Finally, I am infinitely grateful for my brothers and sister Mattias, Felicia, Kenny and Sebastian. And last, but certainly not least, my parents Marianne and Lennart. Thank you for everything. 60 References 1. J.H. Burroughes, D.D.C. Bradley, A.R. Brown, R.N. Marks, K. Mackay, R.H. Friend, P.L. Burns, and A.B. Holmes, Light-emitting diodes based on conjugated polymers. Nature, 1990. 347(6293): p. 539-541. 2. N.S. Sariciftci, L. Smilowitz, A.J. Heeger, and F. Wudl, Photoinduced Electron Transfer from a Conducting Polymer to Buckminsterfullerene. Science, 1992. 258(5087): p. 1474-1476. 3. N.S. Sariciftci, D. Braun, C. Zhang, V.I. Srdanov, A.J. Heeger, G. Stucky, and F. Wudl, Semiconducting PolymerBuckminsterfullerene Heterojunctions - Diodes, Photodiodes, and Photovoltaic Cells. Applied Physics Letters, 1993. 62(6): p. 585-587. 4. Q. Pei, G. Yu, C. Zhang, Y. Yang, and A.J. Heeger, Polymer lightemitting electrochemical cells. Science, 1995. 269(5227): p. 1086-8. 5. A. Sandstrom, A. Asadpoordarvish, J. Enevold, and L. Edman, Spraying Light: Ambient-Air Fabrication of Large-Area Emissive Devices on Complex-Shaped Surfaces. Adv Mater, 2014. 6. C.D. Dimitrakopoulos and P.R.L. Malenfant, Organic Thin Film Transistors for Large Area Electronics. Advanced Materials, 2002. 14(2): p. 99-117. 7. S. Bae, H. Kim, Y. Lee, X. Xu, J.S. Park, Y. Zheng, J. Balakrishnan, T. Lei, H.R. Kim, Y.I. Song, Y.J. Kim, K.S. Kim, B. Ozyilmaz, J.H. Ahn, B.H. Hong, and S. Iijima, Roll-to-roll production of 30-inch graphene films for transparent electrodes. Nat Nanotechnol, 2010. 5(8): p. 574-8. 8. A. Sandstrom, H.F. Dam, F.C. Krebs, and L. Edman, Ambient fabrication of flexible and large-area organic light-emitting devices using slot-die coating. Nat Commun, 2012. 3: p. 1002. 9. M. Hösel, R.R. Søndergaard, D. Angmo, and F.C. Krebs, Comparison of Fast Roll-to-Roll Flexographic, Inkjet, Flatbed, and Rotary Screen Printing of Metal Back Electrodes for Polymer Solar Cells. Advanced Engineering Materials, 2013. 15(10): p. 995-1001. 10. S.R. Forrest, The path to ubiquitous and low-cost organic electronic appliances on plastic. Nature, 2004. 428(6986): p. 911-8. 61 11. P.F. Baude, D.A. Ender, M.A. Haase, T.W. Kelley, D.V. Muyres, and S.D. Theiss, Pentacene-based radio-frequency identification circuitry. Applied Physics Letters, 2003. 82(22): p. 3964. 12. C.C. Chen, L. Dou, R. Zhu, C.H. Chung, T.B. Song, Y.B. Zheng, S. Hawks, G. Li, P.S. Weiss, and Y. Yang, Visibly transparent polymer solar cells produced by solution processing. ACS Nano, 2012. 6(8): p. 7185-90. 13. U. Salzner, J.B. Lagowski, P.G. Pickup, and R.A. Poirier, Comparison of geometries and electronic structures of polyacetylene, polyborole, polycyclopentadiene, polypyrrole, polyfuran, polysilole, polyphosphole, polythiophene, polyselenophene and polytellurophene. Synthetic Metals, 1998. 96(3): p. 177-189. 14. S.S. Lee, J.M. Mativetsky, M.A. Loth, J.E. Anthony, and Y.L. Loo, Quantifying resistances across nanoscale low- and high-angle interspherulite boundaries in solution-processed organic semiconductor thin films. ACS Nano, 2012. 6(11): p. 9879-86. 15. H. Sirringhaus, P.J. Brown, R.H. Friend, M.M. Nielsen, K. Bechgaard, B.M.W. Langeveld-Voss, A.J.H. Spiering, R.A.J. Janssen, E.W. Meijer, P. Herwig, and D.M. de Leeuw, Two-dimensional charge transport in self-organized, high-mobility conjugated polymers. Nature, 1999. 401(6754): p. 685-688. 16. J.-F. Chang, B. Sun, D.W. Breiby, M.M. Nielsen, T.I. Sölling, M. Giles, I. McCulloch, and H. Sirringhaus, Enhanced Mobility of Poly(3-hexylthiophene) Transistors by Spin-Coating from HighBoiling-Point Solvents. Chemistry of Materials, 2004. 16(23): p. 4772-4776. 17. H. Li, B.C.K. Tee, J.J. Cha, Y. Cui, J.W. Chung, S.Y. Lee, and Z. Bao, High-Mobility Field-Effect Transistors from Large-Area SolutionGrown Aligned C60 Single Crystals. Journal of the American Chemical Society, 2012. 134(5): p. 2760-2765. 18. P.C. Eklund, A.M. Rao, P. Zhou, and Y. Wang, Photochemical Transformation of C-60 and C-70 Films. Thin Solid Films, 1995. 257(2): p. 185-203. 19. J.H. Burroughes, C.A. Jones, and R.H. Friend, New semiconductor device physics in polymer diodes and transistors. Nature, 1988. 335(6186): p. 137-141. 62 20. S. Steudel, K. Myny, V. Arkhipov, C. Deibel, S. De Vusser, J. Genoe, and P. Heremans, 50 MHz rectifier based on an organic diode. Nat Mater, 2005. 4(8): p. 597-600. 21. J. Cornil, J.L. Brédas, J. Zaumseil, and H. Sirringhaus, Ambipolar Transport in Organic Conjugated Materials. Advanced Materials, 2007. 19(14): p. 1791-1799. 22. J. Veres, S. Ogier, G. Lloyd, and D. de Leeuw, Gate Insulators in Organic Field-Effect Transistors. Chemistry of Materials, 2004. 16(23): p. 4543-4555. 23. J. Veres, S.D. Ogier, S.W. Leeming, D.C. Cupertino, and S. Mohialdin Khaffaf, Low-k Insulators as the Choice of Dielectrics in Organic Field-Effect Transistors. Advanced Functional Materials, 2003. 13(3): p. 199-204. 24. L.-L. Chua, J. Zaumseil, J.-F. Chang, E.C.W. Ou, P.K.H. Ho, H. Sirringhaus, and R.H. Friend, General observation of n-type fieldeffect behaviour in organic semiconductors. Nature, 2005. 434(7030): p. 194-199. 25. A. Facchetti, M.H. Yoon, and T.J. Marks, Gate Dielectrics for Organic Field-Effect Transistors: New Opportunities for Organic Electronics. Advanced Materials, 2005. 17(14): p. 1705-1725. 26. L. Bürgi, H. Sirringhaus, and R.H. Friend, Noncontact potentiometry of polymer field-effect transistors. Applied Physics Letters, 2002. 80(16): p. 2913. 27. S.M. Sze, Semiconductor Devices, Physics and Technology. 2nd ed. 2002: John Wiley & Sons. 28. C.R. Newman, C.D. Frisbie, D.A. da Silva Filho, J.-L. Brédas, P.C. Ewbank, and K.R. Mann, Introduction to Organic Thin Film Transistors and Design of n-Channel Organic Semiconductors. Chemistry of Materials, 2004. 16(23): p. 4436-4451. 29. J. Zaumseil and H. Sirringhaus, Electron and ambipolar transport in organic field-effect transistors. Chem Rev, 2007. 107(4): p. 1296323. 30. L. Herlogsson, M. Colle, S. Tierney, X. Crispin, and M. Berggren, Low-voltage ring oscillators based on polyelectrolyte-gated polymer thin-film transistors. Adv Mater, 2010. 22(1): p. 72-6. 63 31. A. Dodabalapur, J. Laquindanum, H.E. Katz, and Z. Bao, Complementary circuits with organic transistors. Applied Physics Letters, 1996. 69(27): p. 4227. 32. J.M. Rabaey, A. Chandrakasan, and B. Nikolic, Digital Integrated Circuits, A Design Perspective. Second ed. 2003: Prentice Hall. 33. A. Dodabalapur, Organic and polymer transistors for electronics. Materials Today, 2006. 9(4): p. 24-30. 34. Y.Y. Lin, A. Dodabalapur, R. Sarpeshkar, Z. Bao, W. Li, K. Baldwin, V.R. Raju, and H.E. Katz, Organic complementary ring oscillators. Applied Physics Letters, 1999. 74(18): p. 2714. 35. M. Kitamura, Y. Kuzumoto, S. Aomori, and Y. Arakawa, HighFrequency Organic Complementary Ring Oscillator Operating up to 200 kHz. Applied Physics Express, 2011. 4(5): p. 051601. 36. B. Crone, A. Dodabalapur, Y.Y. Lin, R.W. Filas, Z. Bao, A. LaDuca, R. Sarpeshkar, H.E. Katz, and W. Li, Large-scale complementary integrated circuits based on organic transistors. Nature, 2000. 403(6769): p. 521-3. 37. B.K. Crone, A. Dodabalapur, R. Sarpeshkar, R.W. Filas, Y.Y. Lin, Z. Bao, J.H. O’Neill, W. Li, and H.E. Katz, Design and fabrication of organic complementary circuits. Journal of Applied Physics, 2001. 89(9): p. 5125. 38. D. Bode, K. Myny, B. Verreet, B. van der Putten, P. Bakalov, S. Steudel, S. Smout, P. Vicca, J. Genoe, and P. Heremans, Organic complementary oscillators with stage-delays below 1 μs. Applied Physics Letters, 2010. 96(13): p. 133307. 39. K.-J. Baeg, D. Khim, D.-Y. Kim, S.-W. Jung, J.B. Koo, I.-K. You, H. Yan, A. Facchetti, and Y.-Y. Noh, High speeds complementary integrated circuits fabricated with all-printed polymeric semiconductors. Journal of Polymer Science Part B: Polymer Physics, 2011. 49(1): p. 62-67. 40. R. Chau, S. Datta, M. Doczy, B. Doyle, B. Jin, J. Kavalieros, A. Majumdar, M. Metz, and M. Radosavljevic, Benchmarking Nanotechnology for High-Performance and Low-Power Logic Transistor Applications. IEEE Transactions On Nanotechnology, 2005. 4(2): p. 153-158. 64 41. D.S. Hecht, L. Hu, and G. Irvin, Emerging transparent electrodes based on thin films of carbon nanotubes, graphene, and metallic nanostructures. Adv Mater, 2011. 23(13): p. 1482-513. 42. S. De, T.M. Higgins, P.E. Lyons, E.M. Doherty, P.N. Nirmalraj, W.J. Blau, J.J. Boland, and J.N. Coleman, Silver Nanowire Networks as Flexible, Transparent, Conducting Films: Extremely High DC to Optical Conductivity Ratios. ACS Nano, 2009. 3(7): p. 1767-74. 43. H.A. Becerril, J. Mao, Z. Liu, R.M. Stoltenberg, Z. Bao, and Y. Chen, Evaluation of solution-processed reduced graphene oxide films as transparent conductors. ACS Nano, 2008. 2(3): p. 463-70. 44. J.L. Bredas, J.E. Norton, J. Cornil, and V. Coropceanu, Molecular understanding of organic solar cells: the challenges. Acc Chem Res, 2009. 42(11): p. 1691-9. 45. S. Gunes, H. Neugebauer, and N.S. Sariciftci, Conjugated polymerbased organic solar cells. Chem Rev, 2007. 107(4): p. 1324-38. 46. J.K.J. van Duren, X. Yang, J. Loos, C.W.T. Bulle-Lieuwma, A.B. Sieval, J.C. Hummelen, and R.A.J. Janssen, Relating the Morphology of Poly(p-phenylene vinylene)/Methanofullerene Blends to Solar-Cell Performance. Advanced Functional Materials, 2004. 14(5): p. 425-434. 47. L.J.A. Koster, M. Kemerink, M.M. Wienk, K. Maturová, and R.A.J. Janssen, Quantifying Bimolecular Recombination Losses in Organic Bulk Heterojunction Solar Cells. Advanced Materials, 2011. 23(14): p. 1670-1674. 48. M. Campoy-Quiles, T. Ferenczi, T. Agostinelli, P.G. Etchegoin, Y. Kim, T.D. Anthopoulos, P.N. Stavrinou, D.D. Bradley, and J. Nelson, Morphology evolution via self-organization and lateral and vertical diffusion in polymer:fullerene solar cell blends. Nat Mater, 2008. 7(2): p. 158-64. 49. B. Watts, W.J. Belcher, L. Thomsen, H. Ade, and P.C. Dastoor, A Quantitative Study of PCBM Diffusion during Annealing of P3HT:PCBM Blend Films. Macromolecules, 2009. 42(21): p. 83928397. 50. N.D. Treat, M.A. Brady, G. Smith, M.F. Toney, E.J. Kramer, C.J. Hawker, and M.L. Chabinyc, Interdiffusion of PCBM and P3HT Reveals Miscibility in a Photovoltaically Active Blend. Advanced Energy Materials, 2011. 1(1): p. 82-89. 65 51. G. Lu, L. Li, and X. Yang, Creating a uniform distribution of fullerene C60 nanorods in a polymer matrix and its photovoltaic applications. Small, 2008. 4(5): p. 601-6. 52. Z. Li, H.C. Wong, Z.G. Huang, H.L. Zhong, C.H. Tan, W.C. Tsoi, J.S. Kim, J.R. Durrant, and J.T. Cabral, Performance enhancement of fullerene-based solar cells by light processing. Nature Communications, 2013. 4. 53. H.C. Wong, Z. Li, C.H. Tan, H. Zhong, Z. Huang, H. Bronstein, I. McCulloch, J.T. Cabral, and J.R. Durrant, Morphological stability and performance of polymer-fullerene solar cells under thermal stress: the impact of photoinduced PC60BM oligomerization. ACS Nano, 2014. 8(2): p. 1297-308. 54. M.C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A.J. Heeger, and C.J. Brabec, Design Rules for Donors in BulkHeterojunction Solar Cells—Towards 10 % Energy-Conversion Efficiency. Advanced Materials, 2006. 18(6): p. 789-794. 55. M. Jørgensen, J.E. Carlé, R.R. Søndergaard, M. Lauritzen, N.A. Dagnæs-Hansen, S.L. Byskov, T.R. Andersen, T.T. Larsen-Olsen, A.P.L. Böttiger, B. Andreasen, L. Fu, L. Zuo, Y. Liu, E. Bundgaard, X. Zhan, H. Chen, and F.C. Krebs, The state of organic solar cells—A meta analysis. Solar Energy Materials and Solar Cells, 2013. 119: p. 84-93. 56. A.M. Rao, P. Zhou, K.A. Wang, G.T. Hager, J.M. Holden, Y. Wang, W.T. Lee, X.X. Bi, P.C. Eklund, D.S. Cornett, M.A. Duncan, and I.J. Amster, Photoinduced polymerization of solid C-60 films. Science, 1993. 259(5097): p. 955-957. 57. A. Dzwilewski, T. Wagberg, and L. Edman, Photo-induced and resist-free imprint patterning of fullerene materials for use in functional electronics. J Am Chem Soc, 2009. 131(11): p. 4006-11. 58. J. Wang, C. Larsen, T. Wågberg, and L. Edman, Direct UV Patterning of Electronically Active Fullerene Films. Advanced Functional Materials, 2011. 21(19): p. 3723-3728. 59. T. Wagberg, P.A. Persson, and B. Sundqvist, Structural evolution of low-pressure polymerised C-60 with polymerisation conditions. Journal of Physics and Chemistry of Solids, 1999. 60(12): p. 19891994. 66 60. H.R. Barzegar, C. Larsen, L. Edman, and T. Wågberg, SolutionBased Phototransformation of C60 Nanorods: Towards Improved Electronic Devices. Particle & Particle Systems Characterization, 2013. 30(8): p. 715-720. 61. P.C. Eklund, P. Zhou, K.A. Wang, G. Dresselhaus, and M.S. Dresselhaus, Optical Phonon Modes in Solid and Doped C-60. Journal of Physics and Chemistry of Solids, 1992. 53(11): p. 13911413. 62. B. Burger, J. Winter, and H. Kuzmany, Dimer and cluster formation in C-60 photoreaction. Zeitschrift Fur Physik B-Condensed Matter, 1996. 101(2): p. 227-233. 63. A. Dzwilewski, T. Wagberg, and L. Edman, C-60 field-effect transistors: Effects of polymerization on electronic properties and device performance. Physical Review B, 2007. 75(7): p. 9. 64. A. Dzwilewski, P. Matyba, and L. Edman, Facile fabrication of efficient organic CMOS circuits. J Phys Chem B, 2010. 114(1): p. 135-40. 65. C. Larsen, J. Wang, and L. Edman, Complementary ring oscillator fabricated via direct laser-exposure and solution-processing of a single-layer organic film. Thin Solid Films, 2012. 520(7): p. 30093012. 66. J. Wang, J. Enevold, and L. Edman, Photochemical Transformation of Fullerenes. Advanced Functional Materials, 2013. 23(25): p. 3220-3225. 67. A. Tapponnier, I. Biaggio, and P. Günter, Ultrapure C[sub 60] fieldeffect transistors and the effects of oxygen exposure. Applied Physics Letters, 2005. 86(11): p. 112114. 68. A.L. Briseno, S.C.B. Mannsfeld, S.A. Jenekhe, Z. Bao, and Y.N. Xia, Introducing organic nanowire transistors. Materials Today, 2008. 11(4): p. 38-47. 69. K. Miyazawa, Y. Kuwasaki, K. Hamamoto, S. Nagata, A. Obayashi, and M. Kuwabara, Structural characterization of C60 nanowhiskers formed by the liquid/liquid interfacial precipitation method. Surface and Interface Analysis, 2003. 35(1): p. 117-120. 67 70. K. Miyazawa, Y. Kuwasaki, A. Obayashi, and M. Kuwabara, C(60) nanowhiskers formed by the liquid-liquid interfacial precipitation method. Journal of Materials Research, 2002. 17(1): p. 83-88. 71. C. Park, H.J. Song, and H.C. Choi, The critical effect of solvent geometry on the determination of fullerene (C(60)) self-assembly into dot, wire and disk structures. Chemical Communications, 2009(32): p. 4803-4805. 72. M.G. Yao, B.M. Andersson, P. Stenmark, B. Sundqvist, B.B. Liu, and T. Wagberg, Synthesis and growth mechanism of differently shaped C(60) nano/microcrystals produced by evaporation of various aromatic C(60) solutions. Carbon, 2009. 47(4): p. 1181-1188. 73. A.L. Briseno, S.C.B. Mannsfeld, M.M. Ling, S.H. Liu, R.J. Tseng, C. Reese, M.E. Roberts, Y. Yang, F. Wudl, and Z.N. Bao, Patterning organic single-crystal transistor arrays. Nature, 2006. 444(7121): p. 913-917. 74. K. Ogawa, T. Kato, A. Ikegami, H. Tsuji, N. Aoki, Y. Ochiai, and J.P. Bird, Electrical properties of field-effect transistors based on C-60 nanowhiskers. Applied Physics Letters, 2006. 88(11). 75. M. Larsson, J. Kjelstrup-Hansen, and S. Lucyszyn, DC Characterisation of C60 Whiskers and Nanowhiskers. ECS Transactions, 2007. 2(12): p. 27-38. 76. K.I. Ogawa, N. Aoki, K. Miyazawa, S. Nakamura, T. Mashino, J.P. Bird, and Y. Ochiai, C(60) nanowhisker field-effect-transistor application for nano-electronics. Japanese Journal of Applied Physics, 2008. 47(1): p. 501-504. 77. M.S. Xu, Y. Pathak, D.S. Fujita, C. Ringor, and K. Miyazawa, Covered conduction of individual C(60) nanowhiskers. Nanotechnology, 2008. 19(7). 78. Y. Ochiai, K. Ogawa, N. Aoki, and J.P. Bird, Field-effect-transistor characteristics of solvate C60fullerene nanowhiskers. Journal of Physics: Conference Series, 2009. 159: p. 012004. 79. T. Doi, K. Koyama, Y. Chiba, H. Tsuji, M. Ueno, S.-R. Chen, N. Aoki, J.P. Bird, and Y. Ochiai, Electron Transport Properties in Photo and Supersonic Wave Irradiated C60Fullerene Nano-Whisker FieldEffect Transistors. Japanese Journal of Applied Physics, 2010. 49(4): p. 04DN12. 68 80. K. Miyazawa, Y. Kuwasaki, A. Obayashi, and M. Kuwabara, C60 Nanowhiskers Formed by the Liquid–liquid Interfacial Precipitation Method. Journal of Materials Research, 2011. 17(01): p. 83-88. 81. K. Miyazawa, K. Hamamoto, S. Nagata, and T. Suga, Structural investigation of the C-60/C-70 whiskers fabricated by forming liquid-liquid interfaces of toluene with dissolved C-60/C-70 and isopropyl alcohol. Journal of Materials Research, 2003. 18(5): p. 1096-1103. 82. H.R. Barzegar, F. Nitze, A. Malolepszy, L. Stobinski, C.W. Tai, and T. Wagberg, Water Assisted Growth of C-60 Rods and Tubes by Liquid-Liquid Interfacial Precipitation Method. Molecules, 2012. 17(6): p. 6840-6853. 83. A. Talyzin and U. Jansson, C-60 and C-70 solvates studied by Raman spectroscopy. Journal of Physical Chemistry B, 2000. 104(21): p. 5064-5071. 84. L. Wang, B.B. Liu, S.D. Yu, M.G. Yao, D.D. Liu, Y.Y. Hou, T. Cui, G.T. Zou, B. Sundqvist, H. You, D.K. Zhang, and D.G. Ma, Highly enhanced luminescence from single-crystalline C-60 center dot 1mxylene nanorods. Chemistry of Materials, 2006. 18(17): p. 41904194. 85. D.R. Lide, CRC handbook of chemistry and physics. 2005, Boca Raton: CRC Press. 86. K. Itaka, M. Yamashiro, J. Yamaguchi, M. Haemori, S. Yaginuma, Y. Matsumoto, M. Kondo, and H. Koinuma, High-Mobility C60 FieldEffect Transistors Fabricated on Molecular- Wetting Controlled Substrates. Advanced Materials, 2006. 18(13): p. 1713-1716. 87. S. Kobayashi, T. Takenobu, S. Mori, A. Fujiwara, and Y. Iwasa, Fabrication and characterization of C[sub 60] thin-film transistors with high field-effect mobility. Applied Physics Letters, 2003. 82(25): p. 4581. 88. T.D. Anthopoulos, B. Singh, N. Marjanovic, N.S. Sariciftci, A.M. Ramil, H. Sitter, M. Colle, and D.M. de Leeuw, High performance nchannel organic field-effect transistors and ring oscillators based on C-60 fullerene films. Applied Physics Letters, 2006. 89(21): p. 3. 89. H. Sirringhaus, Device Physics of Solution-Processed Organic FieldEffect Transistors. Advanced Materials, 2005. 17(20): p. 2411-2425. 69 90. L. Edman, J. Swensen, D. Moses, and A.J. Heeger, Toward improved and tunable polymer field-effect transistors. Applied Physics Letters, 2004. 84(19): p. 3744-3746. 91. D.J. Gundlach, L. Zhou, J.A. Nichols, T.N. Jackson, P.V. Necliudov, and M.S. Shur, An experimental study of contact effects in organic thin film transistors. Journal of Applied Physics, 2006. 100(2). 92. L. Burgi, T.J. Richards, R.H. Friend, and H. Sirringhaus, Close look at charge carrier injection in polymer field-effect transistors. Journal of Applied Physics, 2003. 94(9): p. 6129-6137. 93. Y. Liu, C.C. Chen, Z. Hong, J. Gao, Y.M. Yang, H. Zhou, L. Dou, G. Li, and Y. Yang, Solution-processed small-molecule solar cells: breaking the 10% power conversion efficiency. Sci Rep, 2013. 3: p. 3356. 94. J. You, L. Dou, K. Yoshimura, T. Kato, K. Ohya, T. Moriarty, K. Emery, C.C. Chen, J. Gao, G. Li, and Y. Yang, A polymer tandem solar cell with 10.6% power conversion efficiency. Nat Commun, 2013. 4: p. 1446. 95. J. Zhou, Y. Zuo, X. Wan, G. Long, Q. Zhang, W. Ni, Y. Liu, Z. Li, G. He, C. Li, B. Kan, M. Li, and Y. Chen, Solution-processed and highperformance organic solar cells using small molecules with a benzodithiophene unit. J Am Chem Soc, 2013. 135(23): p. 8484-7. 96. J. Peet, J.Y. Kim, N.E. Coates, W.L. Ma, D. Moses, A.J. Heeger, and G.C. Bazan, Efficiency enhancement in low-bandgap polymer solar cells by processing with alkane dithiols. Nat Mater, 2007. 6(7): p. 497-500. 97. W. Li, K.H. Hendriks, W.S.C. Roelofs, Y. Kim, M.M. Wienk, and R.A.J. Janssen, Efficient Small Bandgap Polymer Solar Cells with High Fill Factors for 300 nm Thick Films. Advanced Materials, 2013. 25(23): p. 3182-3186. 98. Z. Li, G.R. He, X.J. Wan, Y.S. Liu, J.Y. Zhou, G.K. Long, Y. Zuo, M.T. Zhang, and Y.S. Chen, Solution Processable Rhodanine-Based Small Molecule Organic Photovoltaic Cells with a Power Conversion Efficiency of 6.1%. Advanced Energy Materials, 2012. 2(1): p. 74-77. 99. E. Ripaud, T. Rousseau, P. Leriche, and J. Roncali, Unsymmetrical Triphenylamine-Oligothiophene Hybrid Conjugated Systems as Donor Materials for High-Voltage Solution-Processed Organic Solar Cells. Advanced Energy Materials, 2011. 1(4): p. 540-545. 70 100. Y. Zhang, Y. Diao, H. Lee, T.J. Mirabito, R.W. Johnson, E. Puodziukynaite, J. John, K.R. Carter, T. Emrick, S.C.B. Mannsfeld, and A.L. Briseno, Intrinsic and Extrinsic Parameters for Controlling the Growth of Organic Single-Crystalline Nanopillars in Photovoltaics. Nano Letters, 2014. 14(10): p. 5547-5554. 101. M. Jorgensen, K. Norrman, S.A. Gevorgyan, T. Tromholt, B. Andreasen, and F.C. Krebs, Stability of polymer solar cells. Adv Mater, 2012. 24(5): p. 580-612. 102. D. Angmo, P.M. Sommeling, R. Gupta, M. Hösel, S.A. Gevorgyan, J.M. Kroon, G.U. Kulkarni, and F.C. Krebs, Outdoor Operational Stability of Indium-Free Flexible Polymer Solar Modules Over 1 Year Studied in India, Holland, and Denmark. Advanced Engineering Materials, 2014. 71