Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Intrinsically disordered proteins wikipedia , lookup
List of types of proteins wikipedia , lookup
Zinc finger nuclease wikipedia , lookup
Protein–protein interaction wikipedia , lookup
Protein mass spectrometry wikipedia , lookup
Structural alignment wikipedia , lookup
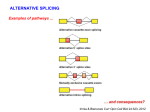
BIPASS: Bioinformatics pipelines alternative splicing services Mentor: Dr. Zoé Lacroix Location: Scientific Data Management Lab. Room GWC 443 The proposed work will contribute to scientific discovery in support of comparative genomics and evolution of proteins; the understanding of the mechanisms underlying genome variation in general and splice site variation in particular will allow the identification of new targets for studying biological systems in humans, model organisms, and microbial environments. In 2004, Lacroix's laboratory at ASU has developed BIPASS a resource that supports alternative splicing events analysis [LLR+07]. BIPASS provides access to transcript sequences retrieved from GenBank that have been pre-processed, clustered with respect to the gene from which they are transcribed, and stored in a database. BIPASS also offers the ability to cluster on-line transcript sequences submitted by scientists. The main findings of the project included (1) the ad-hoc workflow to populate the database did not support updates: the whole database had to be recomputed from scratch each time, (2) this also prevented the ability to combine transcripts submitted by scientists into the existing clusters, (3) the need to use a reference genome without any record of its provenance, the inability to offer real operators to users to query their transcripts against the database. More recent work led to the design of a prediction method to identify alternative splicing events that may lead to mature mRNA that have not yet been observed. This method exploits a partial order to generate a splicing maturity tree from each transcript cluster, as illustrated in Figure 5 [LL08]. Our aim is to exploit the proposed methodology to generate explanations of the consequences of splice-site mutations. Proposed Method for Splice Form Assessment: We will first develop a method for gene discovery and then a technique to support the identification of mutations that may affect splicing, thus protein products. Specifically, given an input sequence (DNA or RNA) and the location of a mutation, we will be able to exploit transcription factors as specific short sequence encoded on specific dimensions and information known about splicing events to (1) identify the signals controlling an encoded gene (here 'signal' refers to a transcription factor as illustrated in Figure 6) and (2) identify the proteins the mutated gene is likely to produce. Moreover, (3) we will infer the properties of these proteins from protein domain classifications. This will exploit both new methodologies developed in our lab. as well as information that is publicly available on the web such as for genomes (UCSC, http://genome.ucsc.edu/), sequences (GenBank, NCBI, http://www.ncbi. nlm.nih.gov/Genbank/), proteins (Swiss-Prot http://www.expasy.ch/sprot/), and known protein classifications (such as SCOP, http://scop.mrc-lmb.cam.ac.uk/scop and Pfam http://pfam. sanger.ac.uk). We will use known promoters including TATA box, cis-trans regulator elements, Ribosomal Binding Site (RBS), enhancers and repressor region, CCAAT-box, GC-box, Downstream Promoter Elements (DPE), Transcription Start Site (TSS+1 site equivalent), Transcription Factor Binding sites (TFBS), B recognition element (BRE), insulators, and initiators [Dat]. These promoters combined with start (ATG) and stop (TAA, TGA, TAG) codons and splicing sites (ESE, ISE, ESS, ISS, and SSM) will constitute the blocks of our method. Indeed we will describe the potential coding regions as words generated by languages such as illustrated by the simplified language AB(CD)+E where A is a transcription site, B is an initiator, C is a translation site, D is a splicing site, and E is a stop codon. The method will be used to predict genes on DNA sequences, and a subsequent alignment with the genome will identify the genes the input sequence originates from. We propose to implement the method with the multidimensional feature extraction algorithms. Unlike traditional alignment methods, they allow the encoding of multiple patterns and the identification of successive patterns in the input sequence. These criteria are critical to the success of the proposed activity. Figure 5. Splicing maturity tree. Figure 6. Language for gene prediction (Eukaryotes only). Proposed Evaluation We will evaluate our proposed methods and algorithms. Gene prediction methods can be evaluated through gene information already published and available. Moreover, we will compare our algorithm against existing tools (benchmark). We will use known splice site mutation events from OMIM to evaluate our splice-site mutation evaluation tool by simulating the effects of splice-site and splice-control site mutations for known cases. We will use simulated datasets to evaluate the prediction methods. However, we will verify manually the results with respect to known published data. We will present and submit papers for peer-review. [LLR+07] Z. Lacroix, C. Legendre, L. Raschid, and B. Snyder, “BIPASS: Bioinformatics pipelines alternative splicing services,” Nucleic Acids Research, vol. 35, web server issue, Suppl. 2, pp. 292-296, July 2007. [LL08] Z. Lacroix and C. Legendre, “BIPASS: Design of alternative splicing services,” International Journal Computational Biology and Drug Design, vol. 1, no. 2, pp. 200– 217, 2008. To apply, a student must contact Dr. Lacroix at [email protected] with a resume upto-date and a letter of motivation (stipulating the reasons why you are interested by the project, if you have prior knowledge of the problems addressed, if you are interested to participate in a cross-disciplinary project, etc.)