Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
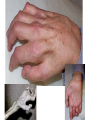
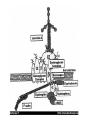
Muscle and neuromuscular junction Neuromuscular diseases (I) Mehmet Ali Akalın V.ENG Neuromuscular Diseases • Group of miscellaneous diseases • Many are hereditary, some are not • Some are chronic, others progress very rapidly • Many share similar symptoms Disorders of the Motor Unit (Classification of Neuromuscular Diseases) • • • • Motor neuron disease Neuromuscular junction disease Muscle disease Peripheral nerve disorders Motor Neuron Disease Diseases that can involve 1) Betz cells of the motor cortex, 2) the lower CN motor nuclei, 3) the CST, 4) and/or the anterior horn cells – Amyotrophic Lateral Sclerosis (ALS) – Progressive bulbar palsy – Progressive muscular atrophy, spinal muscular atrophy – Primary lateral sclerosis ALS • Loss of motor neurons in the cortex, brainstem and spinal cord • Mix of upper motor neuron and lower motor neuron findings – Weakness, atrophy, fasciculations – Slurred speech, difficulty swallowing, shortness of breath • Can start in any extremity or the bulbar musculature • Relentlessly progressive ALS • 50 % dead in 3 years, 80% dead in 5 years, 5-10% live more than 10 years • Death usually from respiratory failure • Etiology still only theoretical – Excess glutamate – Oxidative stress – Free radicals – Mitochondrial dysfunction • There is no therapy to date Myopathies MUSCLE DISEASE Largest group of neuromuscular diseases • Most diverse group • All show a loss of muscle fibers – Proximal more than distal • No involvement of the anterior horn cell, nerve axon, or neuromuscular junction Subcategories 1. 2. 3. 4. 5. 6. Muscular dystrophy Endocrine disorders Metabolic disorders Myotonias Periodic paralysis Polymyositis Endocrine Myopathies • Caused by some malfunction of the endocrine system • Chronic • Respond to drug therapy (primarily hormonal therapy) • Examples: –Addison’s Disease –Cushing’s Syndrome (Steroid myopathy) – Thyrotoxic Myopathy Metabolic Myopathies Myopathies characterized by a deficiency of a specific enzyme resulting in muscle weakness Examples: • McArdle’s Disease – Deficiency of the muscle enzyme myophosphorylase – Glycogen is thus stored in the muscles rather than being used as a source of energy • Pompe’s Disease – deficiency in acid maltase – Excess storage of glycogen in many different organs Case • A 23-year-old man… • was found to have difficulty relaxing after grasping since teenage.. • he has boldness and a thin neck.. • his school performance was poor.. • routine check-up showed cataract and diabetes.. • currently he also has problems with opening eyes and swallowing.. • his mother shares the same disease.. • we heard dive-bomber sound on EMG Myotonias • Characterized by: – Inability to relax a previously contracted muscle • Elicited by either voluntary contractions or some external stimuli such as percussion • Worsened by cold • Lessened by light exercise • Examples: – Myotonic Congenita (Thomsen’s Disease) – Myotonia Atrophica (Myotonic dystrophy) • Myotonic Congenita (Thomsen’s Disease) – Children develop a characteristic hypertrophy of the: • Neck • Deltoid • Biceps • Triceps • Quadriceps, and • Gastrocnemius muscles – Child appears to be a “Tiny Hercules” Myotonic Dystrophy (Steinert’s Disease) – Most frequent neuromuscular disease although it is relatively rare – It is a multisystem disease with major cardiac involvement • Core features of myotonic dystrophy are • myotonia, • muscle weakness, – involvement of the more distal muscles such as: face – neck – Tongue – Intrinsics of hands and feet – • cataract, and • cardiac conduction abnormalities Frontal baldness Myotonic dystrophy (dystrophia myotonica, DM ) • The most frequently inherited neuromuscular disease of adult life • It is a multisystem disease with major cardiac involvement • Core features of myotonic dystrophy are myotonia, muscle weakness, cataract, and cardiac conduction abnormalities Periodic Paralysis • Relatively rare myopathy • Hereditary – Autosomal dominant • Characterized by: – Transient flaccid paralysis or paresis affecting primarily the muscle of the proximal limbs – Attacks of weakness may last from a few seconds to several weeks Primary Periodic Paralyses Sodium channel Hyperkalemic PP Paramyotonia congenita Potassium-aggravated myotonias Calcium channel Hypokalemic PP Potassium channel Andersen-Tawil syndrome Hyperkalemic PP or hypokalemic PP* * Hypokalemic periodic paralysis • where potassium leaks into the muscle cells from the bloodstream. * Hyperkalemic periodic paralysis • where potassium leaks out of the cells into the bloodstream. Hyperkalemic Form – Has increased serum K+ – Triggered by: • Stress • Fasting • Cold • Rest following intensive or prolonged muscular exercise – Attacks minimized by: • Light exercise • Ingestion of carbohydrates Hypokalemic Form – Has decreased serum K+ – Affects men more than women – Triggered by: • Stress • Fasting • Cold • Rest following intensive or prolonged muscular exercise • Alcohol consumption • High carbohydrate diets Polymyositis • Second most common myopathy in adults • Chronic inflammatory condition of muscle • If skin is involved >>>>>(Dermatomyositis) • Insidious onset, Moderately progressive • Clinical signs: – Muscle weakness Flexors more than extensors – Fatigue – – Difficulty swallowing – Joint pain – Mild fever – Weight loss – Very diffuse erythema of face and neck Polymyositis • Presents with proximal muscle weakness in 92% • Myalgias in 25% • Slightly increased risk of cancer – Bladder, lung, lymphoma • Biopsy of muscle confirms diagnosis • Treatment with immunosuppression – Prednisone – Methotrexate Muscular Dystrophy • Largest group of the myopathies • Group of inherited diseases • Characterized by: – Progressive muscle weakness • Examples: – Pseudohypertrophic Muscular Dystrophy (Duchenne’s) – Becker-type Muscular Dystrophy – Facioscapulohumeral Muscular Dystrophy – Limb-girdle Muscular Dystrophy Muscular Dystrophy Duchenne/ Becker Emery-Dreifuss, Congenital Limb-Girdle, Distal Myopathy 2-6 years Childhood to early teens, infancy Late childhood-middle age Life expectancy Rarely beyond 20’s varies Middle age + Inheritance X-linked recessive X-linked recessive, autosomal dom & rec. Autosomal dominant & recessive Dystrophin Emerin, lamin, merosin, etc. Calpain-3, Dysferlin, Caveolin-3, αβδγsargoglycans, etc. Onset Muscle groups affected Genetic linkage Source: www.mdausa.org Dystrophin connects the myofibrils to a complex of proteins in the muscle cell membrane. This in turn connects to the extracellular matrix protein laminin, stabilizing the membrane Case • A school boy , aged 10 yr…with • delayed development of walking since childhood.. • and frequent falls.. • NE showed.. – proximal muscle weakness, waddling gait..toe walking.. – Gower’s sign..lordosis.. – enlarged and stiff calves.. • mental retardation seems obvious. Duchenne's dystrophy (DMD) History and epidemiology • Described in 1852. • The most common X-linked, lethal disease. • Occurs in 1: 3,500 male newborns. Clinical features Skeletal muscle involvement • Onset usually between ages 3 and 5 years. • Proximal muscles and neck flexor muscles (severely) are affected early. • Difficulty doing a sit-up. • Calf hypertrophy (pseudohypertrophy). • Contractures of heel cords and iliotibial bands. • Scoliosis • Cardiomyopathy (can be asymptomatic) • Waddling gait with increased lumbar lordosis. • Difficulty rising from floor; Gower's sign. • Proximal leg muscles are the most severely affected by weakness and wasting. Proximal arm muscles are the next most severely affected. Gower's sign • Cranial nerve supplied muscles are relatively spared. • Between ages 3 and 6 the child's function may improve due to growth and the normal increase in strength, which more than offset the loss of function. • Usually unable to walk by age 10 to 12. • Scoliosis develops following wheelchairdependency. • Death by age 20 in most without a ventilator • Steroids may delay time until wheelchair bound Cardiac involvement • The heart develops fibrosis, mainly in the posterobasal part of the left ventricular wall. • Congestive heart failure and cardiac arrhythmias occur in later stages. • Congestive heart failure may develop in some patients who have adequate respiratory muscle function. Smooth muscle of GI tract • Acute gastric dilatation can cause episodic vomiting, abdominal pain, and gastric distension. • May be mistaken for intestinal obstruction. CNS involvement • The average IQ of DMD patients is one standard deviation below the normal mean. • The intellectual impairment is not progressive. • Verbal IQ is affected more than performance IQ. Case A young man,aged 20 years … was found to have problems with walking since age 12 years.. right now …he has waddling gait.. CK level was as high as 20 times normal.. EMG.. myopathic patterns.. Dystrophin stain showed a defective pattern Becker's dystrophy Clinical features • Onset is usually between age 5 and 15 years. Sometimes, onset is much later. • • By definition, Becker patients walk past the age of 15. • Life expectancy is generally reduced. Other phenotypes • Exertional cramps and myalgia. • Myoglobinuria. • Quadriceps myopathy. Intermediate forms (outliers) • Recognized clinically as early as 3 years. • Preservation of anti-gravity strength in neck flexor muscles. • Stair-climbing and walking to age 12 to 15. • • • • • • A 21-year-old young man …with.. marked contractures of arms and legs..his spine was rigid..he had weakness of biceps and peroneal muscles..he underwent pacemaker implantation because of heart block Emery-Dreifuss Muscular Dystrophy • • • • • • • Genetics ... X-linked recessive ( X q 28 ) emerin defect Features … humeroperoneal weakness plus .. early contractures , rigid spine and heart block Case • • • • • • A 19-year-old male … complaining of progressive worsening of left arm for a couple of years..his left arm shows a Popeye appearance..in addition.. he has difficulty blowing..slight ptosis.. otherwise..he remains quite healthy Facioscapulohumeral Muscular Dystrophy (FSH) • • • • • • Genetics AD , chromosome 4 Features … onset since adolescence slowly progressive , life span unaffected assymmetric weakness and wasting of face,serratus anterior muscle ,biceps muscles,etc. with deltoid spared … Popeye appearance • CK : slightly elevated • EMG : myopathic patterns • Biopsy : myopathic patterns Case • • • • • • • A 52-year-old female …complaining of progressive worsening of weakness and wasting of both legs during the past 6 years..recently she noticed slight weakness in both arms too.. N.E. showed weakness and atrophy of leg and shoulder girdles... Limb Girdle Muscular Dystrophy • Genetics … • heterogenous pathogenesis • Features … • proximal weakness • legs early than arms • face and eye spared • knee jerk diminished early than • ankle jerk Muscular Dystrophy Duchenne/ Becker Emery-Dreifuss, Congenital Limb-Girdle, Distal Myopathy 2-6 years Childhood to early teens, infancy Late childhoodmiddle age Life expectancy Rarely beyond 20’s varies Middle age + Inheritance X-linked recessive X-linked recessive, autosomal dom & rec. Autosomal dominant & recessive Genetic linkage Dystrophin Emerin, lamin, merosin, etc. Calpain-3, Dysferlin, Caveolin-3, αβδγsargoglycans, etc. Onset Muscle groups affected Source: www.mdausa.org Evaluation of the Patient with Suspected Muscle Disease • Lab – Muscle enzymes (CPK, aldolase) – Erythrocyte sedimentation rate (ESR or sed rate) if suspect inflammatory disease – Genetic test • Duchenne’s • Myotonic dystrophy • EMG/NCS • Muscle biopsy • May provide a definitive diagnosis Case for quiz • • • • A 49-year-old mother was examined on the visit…she was found to have lid ptosis, difficulty swallowing and slight weakness in finger flexors and foot drop.. • she also has diabetes and cataract.. • one of her sons shares the same illness... Myasthenia Gravis • An autoimmune disease of the neuromuscular junction caused by autoantibody directed at nicotinic acetylcholine receptors • Can be seen at any age, rare before 10 • Usual Onset : female(20-30 years), male(50-60 years) • F/M = 6/4 • Old/Young = F/M Model of normal neuromuscular junction. Nerve action potential invades the nerve terminal, resulting in enhanced release of acetylcholine. Model of normal neuromuscular junction on left compared with myasthenia neuromuscular junction on the right. Onset Presenting symptoms – – – – Ocular (50%): Ptosis; Diplopia Weakness (35%): Bulbar; Legs; Arms Fatigue (10%) Respiratory failure: Rare Progression: Generally insidious over weeks to months Classification after Osserman & Genkins 1. Adult MG – Group I: Ocular (20%) – Group IIA: Mild generalized (30%) – Group III: Acute fulminating (11%), rapid onset, early respiratory involvement, high mortality. – Group IV: Late Severe (9%), > 2 years after onset. 2. Transient Neonatal MG: 1/6 born to MG mother. Last a few weeks. 3. Congenital Myasthenic Syndrome Symptoms & Signs 1- Weakness • Variable: • increses through the day or • with prolonged physical activity • Onset: • Diplopia or ptosis 2° to extraocular muscle or levator palpebrae weakness • Most patients develop weakness in other muscles • Weakness remains limited to ocular muscles during entire course of the illness myasthenia >>>Ocular A- Ocular weakness • Ptosis & Ophthalmoplegia • Usually asymmetric & bilateral • Pupils: Normal • Rule out focal neural lesions III or VI nerve lesion; Internuclear ophthalmoplegia Especially when unilateral signs B - Facial vweakness : > 95% C- Bulbar weakness Symptoms: Dysarthria, Dysphagia, Weak mastication Signs: – Poor gag reflex & palate elevation; – Weak tongue – May result in aspiration pneumonia Considered life-threatening – Usually an indication for rapidly-acting therapeutic intervention – Plasma exchange most commonly used D- Respiratory muscle weakness Usually due to Diaphragmatic and Intercostal muscle weakness •Strong indication for rapidly-acting therapeutic intervention •Pyridostigmine & Plasma exchange most commonly used May be due to vocal cord paralysis1 •Vocal cords in adductor position: Produces stridor •May require intubation Considered life-threatening E- Systemic muscle weakness Typical: Proximal > Distal; Arms > Legs; Symmetric • Weakness in selective areas • Posterior neck (head ptosis) • Triceps • Quadriceps • Occasionally: Distal musculature Other Symptoms & Signs • 3- Fatigue – Induced by repetitive muscle strength testing or prolonged tonic contraction – Quantitation: Timed upward gaze; Forward arm abduction • 4- Muscle wasting: Uncommon, except when MG is chronic & untreated • 5- Deep tendon reflexes: Usually preserved; May be somewhat brisk in clinically weak muscles • 6- Sensory: Normal Testing • Anti-acetylcholine receptor Ab: – Present in 80% of patient Tindall: – – – – – Ocular 55% positive Mild Generalized 80% positive Moderately severe or acute 100% positive Chronic severe 89% In remission 24% Antibodies to striated muscle (StrAb) • Positive in 30% of all adult onset MG. • Highly associated with thymoma – Positive in 80% of MG patients with thymoma – Positive in 24% of patients with thymoma without MG. – Seronegativity does not exclude thymoma. – Most useful as a marker of thymoma in patients with MG onset before age 40. – A progressive rise in StrAbs titer after resection of thymoma is a good indicator of tumor recurrence. Thymoma • 15% of patient has thymoma, 50% has thymic hyperplasia. • Antiskeletal muscle Ab are detected in 90% of patients with thymoma. • CT Chest detect over 85% of thymoma. • Removal of thymoma produces a delayed improvement of MG 6 - 24 months later. Sustained improvement in > 50%, probably less in older patients. No known long-term side effects. Tensilon test (Edrophonium) Given in incremental doses. Start with 2 mg, observe the response for 45 to 60 seconds, followed by doses of 3 and 5 mg and observation for a clinical response for 1 to 2 minutes following each dose. increased salivation, mild sweating, perioral fasciculations and mild nausea. During the injection, patients often experience 1 to 3 minutes of Hypotension and bradycardia are extremely rare but precautions should be taken. Atropine sulfate (0.6 mg intramuscular or intravenously) should be available in case of an emergency. Tensilon test cont’d • Positive means unequivocally improvement of weakness. • Slight to moderate improvement in muscle strength must be interpreted with extreme caution. • Mild to moderate clinical improvement after tensilon has been reported in: – brain stem lesions, – oculomotor palsy due to cerebral artey aneurysm, – diabetic abducens paresis and – even in normal control subjects. Electrophysiology 1. Repetitive stimulation at 3 hertz. >10% decrement of compound action potential. 2. Single fiber EMG: Increased jitter: jitter is the varying time interval between the triggered muscle action potential in 2 muscle fibers within the same motor unit. • Positive in over 90%. Principles of treatment • Onset before age 60 – Thymectomy, pretreat with plasmapheresis, cholinesterase inhibitors – If response unsatisfactory before or after thymectomy, consider high dose daily prednisone and/or other immunosuppressive agents Principles of treatment • Onset after age 60 – Cholinesterase inhibitors with prednisone, azathioprine or other immunosuppressant – Plasmapheresis for severe exacerbations – Consider thymectomy Principles of treatment • Anticholinesterase useful in all forms • For patient with thymoma, thymectomy is indicated in all ages. They may spread in the mediastinum. • Plasmapharesis is effective, but practical only on a short term basis. Pyridostigmine (Mestinon) Tab: 60 mg, – half life 4 hrs, – take 1 q 4 while awake – Side effect: – diarrhea, – fasciculation, – hypersecretion » treat with Lomotil or Motilium Steroid treatment • Indications: – Insufficient control with Mestinon – Diplopia rarely respond to Mestinon alone – Older male • Start at 100 mg to avoid treatment failure. After remission obtained, switch to alternate day dose, slow taper over 6 to 12 months. Patient may get weaker initially. • Exacerbation is common. Azathioprine (Imuran) – Initial dose 2-3 mg/kg/day. • • • • Complete remission 40%, partial remission 51%, minimal improvement 6.4%, no effect in 2.6%. • Improvement begins in 2-3 months, peaks in 6-15 months. • Keep WBC above 3000/ml. • Monitor liver function weekly X 3 months, then 2x/month. • Sometimes used in combination with steroid. If treatment fails, consider 1. Cyclosporine (Sandimmune) – 5 mg/kg/day, in bid dose. Monitor BP, renal function, Cyclosporine level, Amylase, Cholesterol. – May cause nephropathy, hypertension, hirsutism, liver function abnormality, opportunistic infection, may increase risk of malignancy. 2. Plasma Exchange: short term improvement 3. Human Immune Globulin: Effective but short term improvement Drugs that may adversely affect MG 1. Antibiotics – Aminoglycosides: Neomycin, Gentamycin – Peptide: Polymyxin B, Colistin – Other: tetracycline, Clindamycin, Erythromycin, Ampicillin 2. Neuromuscular blockers: – Botulinum Toxin 3. Cardiac drugs: – Quinine, Quinidine, Procanamide, Lidocaine, Beta blockers, Calcium Channel Blockers Drugs that may adversely affect MG cont’d 4. Miscellaneous: – Epdantion, Oxytocin, Lithium, Magnesium, Diazapam, D penicillamine, Cloroquine, Interferon 5. Corticosteroids may initially produce worsening of MG. Other causes of exacerbation • • • • • Febrile illness Thyroid disease Heat Pregnancy Major Physical Stress