Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Adaptive immune system wikipedia , lookup
Adoptive cell transfer wikipedia , lookup
Gluten immunochemistry wikipedia , lookup
DNA vaccination wikipedia , lookup
Guillain–Barré syndrome wikipedia , lookup
Neuromyelitis optica wikipedia , lookup
Molecular mimicry wikipedia , lookup
Multiple sclerosis research wikipedia , lookup
Immunoprecipitation wikipedia , lookup
Polyclonal B cell response wikipedia , lookup
Immunocontraception wikipedia , lookup
Anti-nuclear antibody wikipedia , lookup
Cancer immunotherapy wikipedia , lookup
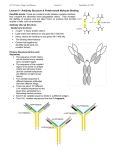
state art nature publishing group Monoclonal Antibody Pharmacokinetics and Pharmacodynamics W Wang1, EQ Wang2 and JP Balthasar3 More than 20 monoclonal antibodies have been approved as therapeutic drugs by the US Food and Drug Administration, and it is quite likely that the number of approved antibodies will double in the next 7–10 years. Antibody drugs show several desirable characteristics, including good solubility and stability, long persistence in the body, high selectivity and specificity, and low risk for bioconversion to toxic metabolites. However, many antibody drugs demonstrate attributes that complicate drug development, including very poor oral bioavailability, incomplete absorption following intramuscular or subcutaneous administration, nonlinear distribution, and nonlinear elimination. In addition, antibody administration often leads to an endogenous antibody response, which may alter the pharmacokinetics and efficacy of the therapeutic antibody. Antibodies have been developed for a wide range of disease conditions, with effects produced through a complex array of mechanisms. This article attempts to provide a brief overview of the main determinants of antibody pharmacokinetics and pharmacodynamics. Introduction Antibodies, which are also called immunoglobulins (Igs), are large proteins used by the immune system to identify and neutralize foreign objects such as bacteria and viruses. All Ig molecules are composed of a basic unit of two identical heavy chains and two identical light chains, held together by a number of disulfide bonds. In humans, there are two types of light chains (κ and λ) and five types of Ig heavy chains (α, δ, ε, γ, and μ).1 Igs are grouped into five classes according to the structure of their heavy chains: IgA, IgD, IgE, IgG, and IgM. Among these, IgG is the predominant class, comprising ~80% of the Igs in human serum. All of the approved therapeutic antibodies are IgGs, and this review focuses on this class. Intact IgGs have a molecular weight of ~150 kDa and a valence of 2 (meaning that each molecule of IgG contains two identical antigen-binding domains). The antigen-binding sites are located in the complementarity determining regions (CDRs) within the Fab portion of the antibody (Figure 1). Fab, which refers to the fragment of antigen binding, is composed of domains associated with the light chain (VL, CL) and domains associated with the heavy chain (VH, CH1). The stem, or Fc, portion of IgG contains the CH2 and CH3 domains of the heavy chains, and this region of the antibody is involved with binding to a wide range of cell-associated receptors (i.e., Fc receptors). The IgG family of antibodies may be further divided, again based on the structure of their heavy chains, into four subclasses: IgG1, IgG2, IgG3, and IgG4. Structural differences among IgG heavy chains lead to differences in subclass binding to Fc receptors and, consequently, to subclass-specific differences in processes mediated by Fc receptors (e.g., activation of complement or antibody-dependent cell-mediated cytotoxicity). For example, antibody-dependent cell-mediated cytotoxicity by mononuclear cells is more efficient for IgG1 and IgG3 than for IgG2 and IgG4. On the other hand, IgG4 is much more active in recruiting the alternative complement pathway than are the other three IgG subclasses.1 Antibody drugs typically possess several desirable pharmacological characteristics, such as long serum half-lives, high potency, and limited off-target toxicity. Initial antibody therapies were prepared from hyperimmune sera, collected following immunization of animals. The resulting antibody product, which is derived from a large number of genetically distinct cells, contains a distribution of Ig isotypes and affinities. In 1975, Köhler and Milstein demonstrated that antibody-producing B lymphocytes may be fused with myeloma cells to generate hybrid cells (hybridomas) that propagate indefinitely in culture and secrete antibody.2 Cloning the hybridoma cells enabled efficient production of antibody derived from a single progenitor, and the resulting monoclonal antibody (mAb) preparations are 1Department of Drug Metabolism and Pharmacokinetics, Merck Research Laboratories, West Point, Pennsylvania, USA; 2Department of Pharmacokinetics, Dynamics, and Drug Metabolism, Pfizer Global Research and Development, Groton Laboratories, Groton, Connecticut, USA; 3Department of Pharmaceutical Sciences, Center for Protein Therapeutics, School of Pharmacy and Pharmaceutical Sciences, University at Buffalo, The State University of New York, Buffalo, New York, USA. Correspondence: JP Balthasar ([email protected]) Received 17 July 2008; accepted 30 July 2008; advance online publication 10 September 2008. doi:10.1038/clpt.2008.170 548 VOLUME 84 NUMBER 5 | NOVEMBER 2008 | www.nature.com/cpt state homogeneous with respect to antibody isotype, primary amino acid sequence, affinity, and specificity. The initial mAbs were generated from mouse and rat hybridomas. These first-generation antibodies found only limited VH VL CL Fab CH1 CH2 Fc CH3 Figure 1 Structure of immunoglobulin gamma (IgG). Four polypeptide chains, including two identical light chains (~25 kDa) and two identical heavy chains (~50 kDa), form the structure of IgG. The light chain contains a variable domain (VL) and a constant domain (CL). The heavy chain is composed of a variable domain (VH) and three constant domains (CH1, CH2, and CH3). The regions associated with antigen binding, or Fab sections of the antibody, include VL, CL, VH, and CH1, whereas the Fc portion of the antibody includes CH2 and CH3. The constant regions are very similar for all IgG antibodies, allowing consistency in structure. Differences between antibodies in the sequence of the variable domains (VL and VH) allow for selective and specific binding of antibodies to different target epitopes on antigens. art success in the clinic because of their short half-lives and high immunogenicity. A number of approaches have been developed to humanize rodent antibodies, from development of chimeric antibodies (where constant regions are typically derived from human IgG, and variable regions are derived from rodent IgG), to “CDR-grafted” antibodies (where CDR regions are derived from the parental rodent IgG, but the remaining 90–95% of the antibody is composed of sequence derived from human IgG), to fully human antibodies. During the past three decades, significant technology advances have been achieved in developing and producing mAbs at commercial scale. To date, the US Food and Drug administration has approved more than 20 mAbs in various therapeutic areas (Table 1). It is estimated that more than 500 mAbs are currently in development. Pharmacokinetic and pharmacodynamic (PK/PD) analyses are essential components of the drug discovery and development process. Antibody drugs often exhibit PK/PD properties that are much more complex than those typically associated with small-molecule drugs (i.e., organic compounds with molecular weight <1,000 Da). Some of the properties had previously been extensively reviewed.3,4 This report provides an overview of the primary determinants of antibody pharmacokinetics and pharmacodynamics, with special attention to state-of-the-art methods of the mathematical modeling of antibody PK/PD. Table 1 Monoclonal antibodies marketed for therapeutic use Antibody Trade name Isotype/structure Primary indication Abciximab REOPRO Chimeric mouse/human Fab Prevention of cardiac ischemic complications Adalimumab HUMIRA Human IgG1 Rheumatoid arthritis Alemtuzumab CAMPATH CDR-grafted rat/human IgG1 B-cell chronic lymphocytic leukemia Basiliximab SIMULECT Chimeric mouse/human IgG1 Prophylaxis of acute organ rejection Bevacizumab AVASTIN CDR-grafted mouse/human IgG1 Colorectal, lung, and breast cancer Certolizumab pegol CIMZIA PEGylated Fab Crohn’s disease Cetuximab ERBITUX Chimeric mouse/human IgG1 Head and neck cancer, colorectal cancer Daclizumab ZENAPAX CDR-grafted mouse/human IgG1 Prophylaxis of acute organ rejection Eculizumab SOLIRIS CDR-grafted mouse/human IgG2/IgG4 Paroxysmal nocturnal hemoglobinuria Efalizumab RAPTIVA CDR-grafted mouse/human IgG1 Psoriasis Gemtuzumab ozogamicin MYLOTARG CDR-grafted mouse/human IgG4 Acute myeloid leukemia Ibritumomab tiuxetan ZEVALIN Murine IgG1 Non-Hodgkin’s lymphoma Infliximab REMICADE Chimeric mouse/human IgG1 Rheumatoid arthritis, Crohn’s disease Muromonab-CD3 ORTHOCLONE OKT3 Murine IgG2a Acute organ rejection Natalizumab TYSABRI CDR-grafted mouse/human IgG4 Multiple sclerosis Omalizumab XOLAIR CDR-grafted mouse/human IgG1 Asthma Palivizumab SYNAGIS CDR-grafted mouse/human IgG1 Prevention of respiratory tract disease Panitumumab VECTIBIX Human IgG2 Colorectal cancer Ranibizumab LUCENTIS CDR-grafted human IgG1 Fab Macular degeneration Rituximab RITUXAN Chimeric mouse/human IgG1 Non-Hodgkin’s lymphoma, rheumatoid arthritis Tositumomab BEXXAR Murine IgG2a Non-Hodgkin’s lymphoma Trastuzumab HERCEPTIN CDR-grafted mouse/human IgG1 Breast cancer CDR, complementarity determining region; IgG, immunoglobulin G. Clinical pharmacology & Therapeutics | VOLUME 84 NUMBER 5 | NOVEMBER 2008 549 state art Antibody Absorption The majority of marketed antibodies are labeled for intravenous (IV) administration; however, several antibodies have been approved for extravascular administration. For example, certolizumab pegol, adalimumab, efalizumab, and omalizumab are all approved for subcutaneous (SC) administration. Palivizumab is approved for intramuscular (IM) administration, and ranibizumab is administered by intravitreal injection. Antibodies have not been successfully developed for oral administration, as oral absorption of antibody is limited by presystemic degradation in the gastrointestinal tract and by inefficient diffusion or convection through the gastrointestinal epithelium. With the exception of ranibizumab, where intravitreal administration is employed to promote a regional effect, efficacy of mAbs following extravascular administration is dependent on systemic absorption. Primary pathways for systemic absorption include convective transport of antibody through lymphatic vessels and into the blood, and diffusion of antibody across blood vessels distributed near the site of injection. Based on work conducted by Supersaxo et al.5 that investigated the lymphatic uptake of a variety of proteins following SC injection in sheep, it has been suggested that the majority of antibody administered via SC or IM injection is absorbed via convection through lymphatic vessels. However, recent investigations conducted in rats suggest that the role of diffusion into blood vessels may be underestimated by the sheep studies.6 Using insulin, bovine serum albumin, and erythropoietin as model proteins, Kagan et al. found that <3% of the administered dose of each protein was absorbed via the lymph. Neither Kagan et al. nor Supersaxo et al. have thoroughly investigated the fate of IgG following SC injection and, consequently, there is substantial uncertainty regarding the primary determinants of antibody absorption. The kinetics of antibody absorption, however, has been well described. After IM or SC injection, absorption proceeds slowly, and the time to reach maximal plasma concentrations (tmax) typically ranges from 2 to 8 days. Absolute bioavailability is generally reported between 50 and 100%.3 In practical terms, bioavailability is determined by the relative rates of presystemic catabolism and systemic absorption. Presystemic catabolism may be dependent on rates of extracellular degradation (e.g., via proteolysis), rates of antibody endocytosis (e.g., receptor-mediated, fluid phase), and rates of recycling through interaction with the Brambell receptor (FcRn). FcRn protects IgG from intracellular catabolism, and FcRn has been shown to be capable of transporting IgG across cell monolayers in both the apical-to-basolateral and basolateralto-apical directions. Work from the Balthasar Laboratory (A. Garg, P.J. Lowe, and J.P. Balthasar, unpublished data) has indicated that the systemic bioavailability of 7E3, a monoclonal IgG1 antibody, was threefold higher in wild-type mice vs. FcRndeficient mice (82.5 ± 15.6% vs. 28.3 ± 6.9%, P < 0.0001). It is not yet known whether the effects of FcRn on SC bioavailability are primarily related to FcRn-mediated protection from catabolism or from FcRn-mediated transport across the vascular endothelium (from interstitial fluid to the blood); however, the former mechanism is considered to be more plausible. 550 In some cases, an inverse relationship between SC bioavailability and antibody dose has been noted.7 Such relationships are suggestive of saturable endocytosis and/or saturable degradation processes. Degradation at the injection site is likely to account for some presystemic loss of antibody, but the quantitative significance is uncertain. Charman et al. have demonstrated that the major determinant of the SC bioavailability of human growth hormone in sheep is presystemic catabolism during the course of lymphatic transport.8 The role of lymphatic catabolism on the bioavailability of other proteins, including mAbs, is not known. As a result of limited solubility of antibodies in solution (~100 mg/ml) and limitations on the volume of fluid that may be tolerated with IM or SC injection (~5 and 2.5 ml, respectively), IM and SC administration are feasible only for antibodies that demonstrate relatively high dose potency. Use of multiple injections may help to overcome this limitation, at least to some extent. For example, doses of 375 mg of omalizumab are routinely administered clinically, via three separate 1-ml SC injections. Although they have not yet been employed in routine clinical use, there is substantial interest in the development of antibodies and Fc-fusion proteins for pulmonary delivery.9 The lungs have a very large surface area and high perfusion rate. In addition, pulmonary epithelial cells are known to express FcRn, which may facilitate efficient systemic absorption of antibody delivered to the lung. As discussed with SC and IM administration, the feasibility of pulmonary delivery of antibodies is likely limited to those antibodies associated with very high dose potency, as only small volumes of fluid may be delivered to the lung. Antibody Distribution The distribution of mAbs is determined by the rate of extravasation in tissue, the rate of distribution within tissue, the rate and extent of antibody binding in tissue, and the rates of elimination from tissue. For large, polar substances such as mAbs, diffusion across vascular endothelial cells is very slow, and convection is believed to be the primary mechanism responsible for the transport of antibody from blood fluid to interstitial fluids of tissue. Of note, physiologically based analyses of antibody disposition in mice suggest that >98% of antibody enters tissue via convection.10 The rate of extravasation by convective transport, or the movement of antibody into tissue by “solvent drag,” is determined by the rates of fluid movement from blood to tissue and by the sieving effect of paracellular pores in the vascular endothelium. Sieving is thought to be largely determined by the size and tortuosity of the pores and by the size, shape, and charge of the solute (i.e., the antibody). Most physiologically based models of antibody disposition describe the uptake clearance for antibody extravasation as a product of the lymph flow rate (L) and an efficiency term (1 − σ). The reflection coefficient, σ, represents the fraction of solute sieved during the movement of solvent through a pore. In the case of mAbs, tissue reflection coefficients are often assumed to be equal in all tissues, with values in the range of 0.95–0.98.10–12 However, it is likely that reflection coefficients may be much lower in tissues such as the VOLUME 84 NUMBER 5 | NOVEMBER 2008 | www.nature.com/cpt state spleen, liver, and bone marrow, where the vascular endothelium is known to be fenestrated or “leaky.” The rate of antibody elimination from tissue will be primarily determined by the convective elimination clearance and by the rates of antibody catabolism within tissue. As with uptake clearance, convective elimination clearance will be a function of the fluid flow rate (i.e., the rate of lymph flow) and sieving. The reflection coefficient associated with convective elimination clearance is related to the diameter of the lymphatic vessels, which carry fluid that drains from the interstitial spaces of tissue. Lymphatic vessels are much larger than paracellular pores in the vascular endothelium and, consequently, it is assumed that there is relatively little restriction of the convective movement of antibody through the lymph. As such, physiologically based models typically assume reflection coefficients of 0–0.2 for convective elimination of antibody via lymphatic drainage. Because of the differences in the efficiency of convective uptake into tissue and convective elimination of antibody from tissue, antibody concentrations in tissue interstitial fluid are substantially lower than antibody concentrations in plasma. In many tissues, concentrations of unbound IgG are approximately tenfold lower than concentrations in plasma; however, higher concentrations are observed in tissues with leaky vasculature (e.g., bone marrow and spleen). IgG antibodies show very little distribution to the brain. The ratio of IgG concentration in the brain relative to plasma is reported to be in the range of 1:500. The poor distribution of antibody to the brain may be explained, in part, by inefficient convective uptake into the brain (e.g., because of the “tight junctions” in the brain vascular endothelium) and by rapid turnover of brain interstitial fluids, which would correlate with efficient convective elimination of IgG from the brain. There is some evidence to suggest that specific Fc receptors, perhaps including FcRn, actively efflux IgG from brain tissue.13 However, investigations conducted in the Balthasar Laboratory have demonstrated virtually identical IgG brain-to-plasma exposure ratios following an IV dose of a murine monoclonal IgG1 antibody in wild-type animals and in FcRn-deficient mice (i.e., 0.0022 ± 0.00015 vs. 0.0021 ± 0.00011, P = 0.3347, A. Garg and J.P. Balthasar, unpublished data). Further investigation is needed to evaluate the possible significance of Fc receptor–mediated efflux of IgG from the brain. Following extravasation, antibody distribution within the interstitial space of tissues is driven by diffusion and convection and, in some cases, restricted as a result of processes of tissue catabolism and cell binding. Some high-affinity mAbs have been reported to show very limited distribution within tissue, where antibody appears to be confined to regions surrounding blood vessels. Nonhomogeneous distribution of antibodies in such tissues has been explained by the “binding-site barrier” hypothesis, which proposes that antibody distribution is restricted because of tight binding to cells near the sites of antibody extravasation.14 The binding-site barrier may be overcome, in theory, with the use of large doses of antibody that may saturate binding sites; however, it may be impossible to administer sufficient doses because of off-target toxicity and/ or feasibility issues. art Analysis of antibody distribution is much more complicated than the analysis of the distribution of most small-molecule drugs. Small-molecule drugs are typically eliminated by the kidney and liver. As a result of the high rate of perfusion of these organs, and the high tissue permeability of most small-molecule drugs, concentrations of drug at the site of drug elimination (i.e., liver and/or kidney) appear to be in very rapid equilibrium with the concentration of drug in plasma. For such drugs, the apparent volume of distribution at steady state (Vss) is independent of the rate of elimination clearance, and Vss may be inferred from plasma data through the use of standard noncompartmental analyses or through data-fitting with mammillary compartmental models. For macromolecular protein drugs, it is possible, and perhaps likely, that a significant fraction of drug elimination occurs from tissue sites that are not in rapid equilibrium with plasma. In such situations, noncompartmental analysis of plasma data will lead to an underestimation of Vss.3 In cases where antibody shows high-affinity, high-capacity binding in tissue, and “target-mediated elimination,” the true Vss may be more than tenfold greater than the distribution volume estimated by standard noncompartmental analyses. When significant drug elimination occurs from “peripheral compartments,” it is not possible to obtain precise estimates of Vss from analysis of plasma data alone; however, Mordenti and Rescigno have shown that, in certain situations, plasma data may be used to define the range of possible values for the distribution volume.15 Precise analysis of Vss requires concentration data from both plasma and tissues. Antibody Elimination Common mechanisms of drug elimination include filtration (e.g., into urine), secretion (e.g., into the bile), and biotransformation (e.g., metabolism or catabolism). Renal elimination, which is a primary pathway of clearance of small-molecule drugs, is relatively unimportant for IgG, as its large size prevents efficient filtration through the glomerulus. Secretion into the bile is an important pathway of elimination of IgA antibodies, but this route is not a significant contributor to the elimination of IgG antibodies. The majority of IgG elimination occurs via intracellular catabolism, following fluid-phase or receptormediated endocytosis. Receptor-meditated endocytosis of IgG may proceed following interaction of the Fab binding domains of the antibody with target epitopes found on the cell surfaces. This type of endocytosis and elimination is a form of target-mediated disposition where the interaction of the drug and its pharmacological target (e.g., a target receptor) serves as a significant contributor to the kinetics of antibody distribution and elimination. Target-meditated elimination is, by definition, capacity limited (saturable) because of finite expression of the target. The rate of uptake and elimination of antibodies by target-mediated pathways is a function of dose and the expression level of the target, as well as a function of the kinetics of receptor internalization and intracellular catabolism. It is important to note, however, that target-mediated elimination does not necessarily require Fab binding to a cell-surface receptor. Certain soluble substances, particularly multimeric Clinical pharmacology & Therapeutics | VOLUME 84 NUMBER 5 | NOVEMBER 2008 551 state art substances with several repeated epitopes, may bind with two or more antibodies, leading to the formation of large complexes that may be rapidly eliminated by phagocytosis. Elimination of large immune complexes may explain, in part, the nonlinear elimination kinetics of omalizumab and denosumab, which are thought to interact with soluble targets (IgE and receptor activator of nuclear factor-κB ligand). The majority of marketed antibodies demonstrate dose-dependent elimination consistent with target-mediated elimination, where clearance decreases as a function of dose (e.g., trastuzumab, rituximab, gemtuzumab, and panitumumab). Examples of state-of-the-art mathematical modeling of target-mediated antibody elimination include reports by Ng et al., describing the nonlinear disposition of TRX1, an anti-CD4 mAb;16 by Hayashi et al., describing the nonlinear disposition of omalizumab, an anti-IgE mAb;17 and by Lammerts van Bueren et al., presenting an interesting conceptual model of target-mediated antibody elimination from a peripheral distribution compartment.18 IgG antibodies may also interact with Fc-γ-receptors (FcγR), and IgG-FcγR complexes may trigger endocytosis and catabolism. Considering the relatively high affinity of IgG for FcγR and the high endogenous concentrations of IgG in plasma (~65 μmol/l), it has been argued that FcγR-mediated elimination is unlikely to be important for monomeric IgG.3 It is possible that FcγR-mediated elimination is significant, and perhaps dominant, in cases where antibody is able to form soluble immune complexes containing three or more IgG molecules, as well as in cases where antibody binds to cells suspended in blood or other body fluids (perhaps including viruses, bacteria, platelets, erythrocytes, and leukocytes). IgG “opsonized” particles are rapidly engulfed following engagement of FcγR on macrophages and on other phagocytic cells. This mechanism of elimination is well supported by the immunology literature; however, little work has been performed to link FcγR-mediated phagocytosis to the systemic pharmacokinetics of therapeutic antibodies. Additional study is required to allow meaningful discussion of the role of FcγR-mediated endocytosis in the elimination of such antibodies. IgG, like other proteins found in plasma and interstitial fluid, may enter cells in all tissues via fluid-phase endocytosis. Interestingly, however, IgG differs from most proteins in that a significant fraction of endocytosed IgG is not sorted to the lysosome but is redirected to the cell surface and released into plasma or interstitial fluids. The recycling of IgG is mediated by the Brambell receptor, FcRn, which binds to IgG with pHdependent affinity.19,20 Within the acidified environment of the early endosome, IgG binds tightly to FcRn. The IgG–FcRn complexes are not delivered to the lysosome for catabolism but rather are sorted to the cell surface for fusion with the cell membrane. The receptor shows virtually no affinity for IgG at physiological pH and, upon fusion of the sorting vesicle with the cell membrane, IgG dissociates from the receptor and is rapidly released into extracellular fluid. FcRn-mediated recycling of IgG appears to be quite efficient based on studies conducted with knockout mice. In animals lacking expression of FcRn, IgG clearance is increased 552 by approximately tenfold,20 which would be consistent with a recycling efficiency of 90% (i.e., in wild-type animals expressing FcRn). Because FcRn expression is limited, FcRn-mediated recycling is capacity limited. The average concentration of IgG in plasma in humans is ~10 mg/ml. At this concentration, IgG has a half-life of ~25 days21 and a plasma clearance of ~10 ml/h (~3.5 ml/kg/day). High concentrations of IgG are able to saturate the recycling system, decreasing recycling efficiency and leading to an increase in the fractional catabolic rate of IgG. For example, in myeloma patients, where IgG concentrations in plasma may approach 100 mg/ml, IgG half-life decreases to 8–10 days. Conversely, in patients with very low plasma concentrations of IgG, the half-life of IgG antibody may be >70 days.21 IgG affinity for FcRn is species specific. Human FcRn shows high affinity for human IgG and also for IgG from guinea pigs and rabbits; however, the human receptor shows very little affinity for IgG derived from most other species, including mice and rats.22 The low affinity of human FcRn for mouse IgG helps to explain the very rapid elimination of murine mAbs in humans. Approved murine monoclonal IgGs (e.g., muromononab-CD3, ibritumomab) demonstrate half-lives of ~1 day in patients, whereas human IgG is typically associated with a half-life of ~25 days. Although FcRn recycling is capacity limited, significant alteration in the efficiency of FcRn recycling is not typically achieved with therapeutic doses of mAbs. Most mAbs are administered at doses of <10 mg/kg, which will increase the total IgG “body load” by <1–2%, as humans typically possess 50–100 g of endogenous IgG. However, high-dose intravenous immunoglobulin (IVIG) therapy, which calls for the administration of 2 g/kg of pooled human IgG, increases IgG plasma concentrations sufficiently to increase IgG clearance approximately threefold.23 This increase in IgG clearance leads to a decrease in endogenous antibody concentrations; consequently, IVIG therapy for the treatment of autoimmune conditions may achieve effects by decreasing the plasma concentrations of endogenous, pathogenic autoantibodies. Although IVIG therapy is an effective treatment of a variety of autoimmune conditions, it is very expensive because of the high doses of antibody required. Of note, preclinical experiments have demonstrated that anti-FcRn antibodies are able to achieve effects similar to those of IVIG therapy, at dose levels that are ~100-fold lower than those required for use in IVIG therapy.24 There is significant interest in the development of specific FcRn inhibitors for use in the treatment of autoimmunity.25 Immunogenicity Any exogenous protein may be viewed by the body as foreign and trigger immune responses that lead to the generation of endogenous antibodies against the protein. Therapeutic antibodies are no exception. mAb drugs may be categorized as (i) rodent antibodies, which are typically obtained from murine or rat hydridomas; (ii) chimeric antibodies, which are derived from chimeras that have been engineered to express IgG antibodies with human constant regions and rodent variable regions; (iii) CDR-grafted antibodies, which contain specific regions within rodent variable domains, the CDRs, grafted onto a human IgG VOLUME 84 NUMBER 5 | NOVEMBER 2008 | www.nature.com/cpt state framework; and (iv) antibodies that are fully human. In chimeric antibodies, ~67% of the primary sequence of the antibody is derived from the human sequence, and ~33% is derived from the rodent sequence. In the case of CDR-grafted antibodies, ~95% of the primary sequence is derived from a human IgG, and ~5% is derived from a rodent antibody. Immunogenicity (i.e., the ability of a protein to induce immune response) is associated with the fraction of foreign sequence in the therapeutic antibody.26 For example, when immunogenicity of a CDR-grafted form of anti-Tac antibody and its murine counterpart were compared in cynomolgus monkeys, it was found that monkeys treated with the humanized version exhibited five- to tenfold lower anti-drug antibody titers.27 Early rodent mAbs were shown to be highly immunogenic in humans. Muromonab-CD3 (OKT3), the first murine mAb approved by the Food and Drug administration, has been associated with high incidences of anti-drug antibodies that interfere with its function.28 In contrast, chimeric, humanized, and fully human therapeutic antibodies are associated with much lower frequency of immunogenicity, ranging from <1% to ~10% in most cases. Of note, however, assays for immunogenicity are difficult to develop and verify, and the direct comparison of immunogenicity results is complicated by potential differences in assay sensitivity.28 Nevertheless, it is fair to say that all therapeutic antibodies currently on the market have shown at least some immunogenicity. Even fully human antibodies have unique idiotypes and, in some cases, unique posttranslational modifications or impurities associated with the manufacturing process that may trigger an immune response. Other factors associated with immunogenicity include duration of therapy, dose, and route of administration.26 Perhaps as expected, the degree of immunogenicity increases with duration of therapy. In the case of infliximab, a chimeric antitumor necrosis factor antibody, anti-drug antibodies are infrequently detected within the first 2 months of therapy; however, after 12 months of therapy, anti-drug antibodies are found in >90% of treated patients.29 Interestingly, anti-drug antibodies are found more frequently, in many cases, after low-dose therapy vs. after higher doses of therapeutic antibody. For example, Stephens et al. demonstrated that after administration of the antibody CDP571 to human subjects at doses ranging from 0.1 to 5 mg/kg, anti-CDP571 titers decreased with increasing dose.30 This phenomenon, which has been reported for other therapeutic antibodies, may reflect an actual inverse relationship between immunogenicity and dose; however, the data may also be explained by assay interference (i.e., where the presence of higher quantities of drug in the sample affects the ability of the assay to detect anti-drug antibodies). Several reports suggest that administration of biologics by the SC and IM routes leads to greater immunogenicity than that observed following the IV route; however, this has not been convincingly demonstrated in humans. Immunogenicity can affect the safety, pharmacokinetics, and pharmacodynamics of therapeutic antibodies. The clinical significance of immunogenicity is product specific and has been reviewed extensively elsewhere.31 The presence of anti-drug antibodies may lead to a wide range of PK effects, and the effect art of endogenous, anti-drug antibodies on a given exogenous protein may depend on the number of antigenic sites found on the exogenous protein. In cases where only one or two endogenous anti-drug IgG molecules bind to the exogenous protein, the halflife of the exogenous protein may actually increase and approach that of endogenous IgG. On the other hand, in cases where three or more IgGs bind to the exogenous protein simultaneously, the resulting immune complex will be eliminated very rapidly through phagocytosis.32 The impact of endogenous anti-drug antibodies on the disposition of therapeutic antibodies may be complex and difficult to predict. However, it is reasonable to expect that, with increased exposure to a therapeutic antibody, there is an increasing probability of development of endogenous antibodies against multiple antigenic sites, as well as an increasing probability that endogenous antibodies will mediate rapid elimination of the therapeutic antibody. Interspecies Scaling In many cases, PK parameters of small-molecule drugs may be scaled across species, via the principles of allometry, with reasonable precision. Such scaling is often accomplished using a simple power model of the form Y = a BWb, where Y is the parameter of interest, BW is the body weight, a is the allometric coefficient, and b is the allometric exponent. Allometric scaling was first applied to proteins by Mordenti et al. in 1991.33 In their work, the authors applied power models to five therapeutic proteins, including a fusion protein composed of CD4 and the Fc portion of an IgG1 molecule. It was shown that the clearance and volume data from preclinical species can be satisfactorily described by the power equation, and the predictions of clinical parameters were very close to the observed values. Interestingly, exponent values for clearance and volume were close to the “expected” values of 0.75 and 1 for small-molecule drugs. Other examples of successful application of allometric scaling to proteins and antibody drugs include work by Grene-Lerouge et al. for antibody fragments,34 Woo and Jusko (2007) for human recombinant proteins (erythropoietin),35 and Vugmeyster et al. for mAbs.36 Although these reports suggest that allometric power relationships may be used to predict clinical antibody pharmacokinetics, one must proceed with caution. Assumptions underlying allometric scaling include the absence of nonlinear pharmacokinetics and species-specific clearance, which may not hold true for most therapeutic antibodies. For example, in an unsuccessful attempt, allometric scaling failed to predict clearance of a murine antiEGF/r3 mAb in cancer patients.37 This is likely because of both the low affinity of murine antibodies for human FcRn and the increased target-mediated clearance in cancer patients. Another approach for predicting drug pharmacokinetics in humans based on preclinical data is to employ physiologically based PK (PBPK) modeling. PBPK models represent the body with several interconnected compartments, each representing an organ. The size of each compartment is based on the physical size of distribution spaces within the organ of interest, and the intercompartmental transfer functions relate to physiological processes (e.g., blood perfusion and lymph flow). Data obtained in preclinical species, together with actual physiological parameters Clinical pharmacology & Therapeutics | VOLUME 84 NUMBER 5 | NOVEMBER 2008 553 state art such as volume of each compartment (organ) and blood flow rate, are used to build the model and predict human pharmacokinetics. A significant advantage of the PBPK approach is that it allows prediction of antibody levels in many tissues, including tumor. PBPK models are ideally suited to the consideration of effects of saturable processes (e.g., target binding, FcRn processing) on antibody pharmacokinetics, and these models are also well suited to predict the influence of a variety of factors (e.g., antigen expression, antibody affinity) on the tissue selectivity of antibody disposition. Recent PBPK models have incorporated FcRn-antibody binding, allowing consideration of the effects of FcRn on antibody catabolism and distribution.11,12 The limitations for use of PBPK models to predict the disposition of antibodies in humans are significant, however. PBPK models are complex, mathematically difficult to construct, poorly suited to population analyses, and often limited because of a lack of tissue concentration data, parameter availability, or parameter identifiability. Despite the large number of antibodies in development, only a handful of reports using preclinical data to predict the clinical pharmacokinetics of antibodies have been published, perhaps indicating the difficulties associated with the interspecies scaling of antibody disposition. Any effort to predict human pharmacokinetics based on preclinical disposition data should consider possible species differences in the expression or turnover of the target receptor, antibody affinity for the target, antibody-FcRn binding, endogenous IgG concentrations (i.e., as a determinant of FcRn saturation), and potential effects of host anti-drug antibodies. Pharmacodynamics mAbs have been marketed for use in the treatment of a wide range of conditions, including cancer, autoimmunity, and inflammatory diseases. It is convenient to discuss antibody pharmacodynamics relating to four main categories of applications: (i) immunotoxicotherapy, where antibody is employed to alter the pharmacokinetics and pharmacodynamics of soluble ligands (e.g., drugs, xenobiotics, and cytokines); (ii) elimination of target cells; (iii) alteration of cellular function (e.g., receptor blockade); and (iv) targeted drug delivery.3 Antibodies used for immunotoxicotherapy include bevacizumab, adalimumab, ranibizumab, omalizumab, and infliximab. Each of these antibodies binds to a soluble ligand (e.g., vascular endothelial growth factor or tumor necrosis factor) and alters the pharmacokinetics and pharmacodynamics of the ligand. These “neutralizing” antibodies act as competitive inhibitors of ligand-receptor binding, shifting ligand concentration–effect relationships. In addition, by binding to soluble ligand, immunotoxicotherapies often produce dramatic alterations in ligand pharmacokinetics. In most cases, the anti-ligand antibody will decrease the unbound fraction of ligand in plasma, decrease the ligand volume of distribution and clearance, and increase the halflife of the ligand. For example, omalizumab, an anti-IgE mAb, dramatically decreases the clearance of its target ligand, leading to a fivefold increase in the plasma half-life of IgE. PK/PD models for omalizumab and infliximab have been published recently.38,39 In each model, a second-order association 554 function was employed to describe the formation of antibody– ligand complexes, and complexes dissociated via a first-order process. The models are nonlinear with respect to antibody– ligand binding because of the second-order nature of the binding process, as well as capacity limitations associated with the available concentrations of ligand and antibody. The models relate unbound ligand concentration to the effect of interest; as such, the models link the PK effects of the anti-ligand antibody to the pharmacodynamics of the ligand. Of note, the models differ substantially in terms of their characterization of the fate of the antibody–ligand complex. In the infliximab model, it was assumed that the antibody–ligand complex was eliminated with the same fractional catabolic rate as the unbound ligand, tumor necrosis factor-α. Fitting the model parameters to the data resulted in an estimation of a 30–40-day half-life for tumor necrosis factor-α, which is considerably different from the known value (<1 h). The omalizumab model, which is much more plausible, does not assume an equivalent elimination rate constant for the complex and the ligand (IgE) but allows for kinetically distinct elimination of IgE, omalizumab, and the IgE–omalizumab complex. The recent work of Marathe et al., which describes the PK/ PD of denosumab, a monoclonal IgG2 antibody directed against the receptor activator of nuclear factor-κB ligand, represents the state of the art in modeling immunotoxicotherapies (Figure 2).40 The receptor activator of nuclear factor-κB ligand is thought to be a soluble ligand, but there is some possibility that the protein is also expressed on the cell surfaces. Denosumab pharmacokinetics were captured with a targetmediated disposition model, and denosumab pharmacodynamics were described with a model that relates the unbound concentrations of denosumab to the inhibitory effect of the antibody on the receptor activator of nuclear factor-κB ligand binding. This mechanistic model provided an excellent description of the pharmacokinetics and pharmacodynamics of denosumab in multiple myeloma patients. Several antibodies, including rituximab, cetuximab, and trastuzumab, are designed to bind to cell-surface proteins to mediate Osteoclast precursors Denosumab–RANKL Active osteoblasts RANKL RANK–RANKL Active osteoclasts OPG–RANKL Serum NXT Figure 2 Pharmacodynamic model for denosumab. Marathe et al. provide an excellent example of a pharmacodynamic model for an antibody acting as an antagonist of a soluble ligand.40 Denosumab, like other antibodies used for immunotoxicotherapy, binds to a soluble ligand (receptor activator of nuclear factor-κB ligand, RANKL), preventing the ligand from binding to its endogenous receptor (receptor activator of nuclear factor-κB, RANK), and antagonizing the effect of the ligand (i.e., inhibiting RANKL stimulation of osteoclast maturation). The Marathe et al. model, which has been simplified herein, employs equilibrium binding functions to relate plasma concentrations of denosumab, RANK, and the natural RANKL antagonist (osteoprotegrin, OPG) to unbound concentrations of RANKL, and to the measured biomarker (serum N-telopeptide, NTX). VOLUME 84 NUMBER 5 | NOVEMBER 2008 | www.nature.com/cpt state the destruction of target cells. Antibodies may eliminate cells by blocking or cross-linking cellular receptors and inducing apoptosis or by “effector functions” of the immune system (e.g., the classical complement pathway, FcγR-mediated phagocytosis). The efficacy of an IgG antibody in mediating cellular destruction is determined by several factors, including the antibody isotype, the expression of the target protein on the cell surface, the signaling pathways associated with the target protein, and the availability of complement proteins (e.g., C1q) and effector cells (e.g., macrophages). Variation in target expression or in the expression of receptors associated with effector pathways may be expected to lead to interindividual variability in antibody pharmacodynamics. Indeed, the efficacy of anti-CD20 antibodies (e.g., rituximab) is significantly different in patients expressing FcγRIIIa with valine vs. phenylalanine at position 158 in the primary sequence of the receptor.41 Although a wealth of information is available in the immunology literature that describes the relationships between effector components and antibody-dependent cell cytotoxicity, little work has been undertaken to incorporate this information within mechanistic PK/PD models. There has been little modeling of the pharmacodynamics of marketed antibodies that mediate cell destruction. Meijer et al. have presented a mechanistic model describing the target-mediated disposition of an anti-CD3 antibody, including characterization of the time course of CD3 in the plasma of treated patients,42 and Mould et al. have presented a semi-mechanistic PK/PD model describing the effects of anti-CD4 mAb administration on CD4-positive T cells in patients with rheumatoid arthritis.43 PD models have not been published, to the authors’ knowledge, for rituximab, trastuzumab, or cetuximab. However, several reports have described the PK/PD modeling of preclinical data. For example, Sharma et al. developed an indirect effect model to describe the pharmacodynamics of keliximab and clenoliximab, which are monkey/human chimeric antibodies directed against CD4.44 Although keliximab and clenoliximab have identical variable domains, keliximab is an IgG1 and clenoliximab is an IgG4. Relative to IgG4 antibodies, IgG1 antibodies demonstrate greater potential for antibody-dependent cell cytotoxicity and activation of complement by the classical pathway. Consequently, keliximab would be expected to be more efficient in eliminating CD4-positive T cells. Consistent with this expectation, the PD modeling demonstrated that keliximab enhanced the elimination of CD4-positive T cells with much greater potency than did clenoliximab. The fit value for SC50, which refers to the antibody concentration leading to half-maximal effect, was tenfold lower for keliximab vs. clenoliximab. Similar models have been developed to describe the pharmacodynamics of anti-platelet antibodies in mouse and rat models of immune thrombocytopenia.45 The Deng model is shown in Figure 3 as an example of characterization of the pharmacodynamics of antibodies used to stimulate the elimination of target cells. Abciximab, basiliximab, daclizumab, and efalizumab are examples of marketed antibodies that achieve effects by altering cell signaling. In addition, some of the effects induced by rituximab and cetuximab are associated with their effects on signaling pathways. Abciximab is a Fab fragment that binds to the GPIIb/ I3 τ I2 kin τ PLT I1 τ IPLT x art PLT −R0 R0 kout APAb IVIG CL Figure 3 Pharmacodynamic model for anti-platelet antibodies. The Deng et al. model, selected as an example model for antibodies that mediate the elimination of cells, utilizes an indirect response function to describe the stimulatory effect of anti-platelet antibodies on the elimination of platelets. Platelet production proceeds by a cascade of events (e.g., megakaryoctye maturation), and this is captured through the use of a transduction function, where τ represents the time delay associated with each step in the maturation process. Platelet count provides feedback inhibition on platelet production (I3, kin). Intravenous immunoglobulin (IVIG) treatment leads to a stimulation of anti-platelet antibody clearance (CL) via saturation of FcRn and also leads to an inhibition in the elimination (kout) of opsonized platelets. Figure adapted from ref. 45. IIIa receptor on platelets. By binding to the receptor, abciximab competitively inhibits platelet binding to fibrinogen and the von Willebrand factor, thereby inhibiting platelet aggregation. Abciximab, as a Fab fragment, does not possess the Fc-domains required for mediating antibody-dependent cell cytotoxicity, and, consequently, abciximab does not typically induce thrombocytopenia in patients. Intact anti-GPIIb/IIIa antibodies, on the other hand, lead to significant thrombocytopenia in animal models. Mager et al. have developed an inhibitory Emax model to describe the effects of abciximab on ex vivo platelet aggregation, using data obtained from patients undergoing coronary angioplasty.46 The PD model, which assumed that abciximab concentrations in plasma are directly related to abciximab effects, provides excellent characterization of the data and serves as a good example for use in the analysis of “direct effects” of antibody drugs. Efalizumab binds to CD11a, which is a subunit of leukocyte function antigen-1, and triggers a decrease in the cellular expression of the receptor. By decreasing receptor expression and by binding to and occupying the available CD11a, efalizumab is an effective inhibitor of CD11a-mediated cell signaling. The effects of efalizumab on CD11a expression and on CD3-positive lymphocytes in chimpanzees and in psoriasis patients have been captured with mechanistic PK/PD models.47 Two models were developed for analysis of data collected from chimpanzees, each of which utilized an indirect response relationship to describe CD11a turnover. Although the models differed in terms of their characterization of the pathways associated with the nonlinear elimination of the antibody, each model provided very good characterization of the PK/PD data. Subsequent dose-ranging studies were conducted in psoriasis patients, and a population PK/PD approach was used to characterize the data using each model. Again, each model was able to capture the data well. More recently, Ng et al. reported a mechanistic PK/PD model in which the authors characterized the kinetic relationships between plasma efalizumab exposure and efalizumab effects on CD11a expression on T cells and on Clinical pharmacology & Therapeutics | VOLUME 84 NUMBER 5 | NOVEMBER 2008 555 state art Vm · efalizumab·CD11a Vc · Km + efalizumab CD11a CD11a production kel kPASI PASI kheal Figure 4 Pharmacodynamic model for efalizumab. The Ng et al. model, selected as an example model for antibodies that alter cellular function, describes the effects of efalizumab on CD11a expression, elimination, and the psoriasis area and severity index (PASI). The model allowed excellent characterization of efalizumab pharmacodynamics in a large, population pharmacokinetic and pharmacodynamic analysis. The figure shown is a simplified version of the model presented by Ng et al.48 the psoriasis area and severity index, an efficacy end point for psoriasis (Figure 4).48 Preclinical modeling examples include a report by Luo et al. that describes the development of a model of cetuximab PK/ PD using data collected from studies conducted with a murine colon carcinoma xenograft model.49 Although the antibody demonstrates nonlinear, target-mediated disposition in humans, cetuximab pharmacokinetics were dose-proportional in the mouse model and were well characterized with a linear, onecompartment model. Cetuximab’s effects on the phosphorylation of the epidermal growth factor receptor were captured with an indirect effect model, which allowed comparison between estimated values of EC50 and EC90 (half maximal effective concentration and 90% effective concentration, respectively), plasma concentrations of cetuximab achieved in patients, and the efficacy of cetuximab in clinical trials. In comparison with the other main categories of antibody usage, relatively little success has come from the development of antibodies for targeted drug delivery. Most of the interest in this area has centered on the development of conjugates of antibodies and toxic agents (e.g., chemotherapeutic drugs, radioisotopes, and biological toxins), with the intent of using the high specificity and selectivity of antibodies to mediate targeted delivery of toxins. The antibody–toxin conjugates, or immunotoxins, carry the complexities shared by other types of antibody drugs (e.g., potential for nonlinear target-mediated disposition, immunogenicity). In addition, off-target toxicity is often a greater concern for immunotoxins because of the potential for dissociation, in vivo, of the toxin from the antibody and because of the high potency of toxins employed. Considerable toxicity often results from “nonspecific” distribution of the immunotoxin to off-target sites. Bone marrow stem cells are particularly susceptible to toxicity from immunotoxins because of their rapid growth rate and high sensitivity to chemotherapy, along with the leaky vasculature of the bone marrow, which allows relatively efficient convective uptake of immunotoxins. 556 Most of the work associated with the use of antibodies for targeted drug delivery has been focused on the treatment of solid tumors. Solid tumors are problematic targets for antibody drugs, as tumor growth often leads to the collapse of lymphatic vessels within the tumor, which leads to an increase in the tumor interstitial pressure. High interstitial pressure minimizes the blood-to-tumor hydrostatic pressure gradient, and this decreases the driving force for antibody uptake into tumor by convection. Once antibody extravasates, distribution may be limited by the binding-site barrier (discussed above), further reducing the effectiveness of antibody-directed delivery of toxins to solid tumors. For chronic immunotoxin therapy, it may be important to select a cellular target that is easily accessed by antibody in blood (i.e., hematological cells, cells in tissues with “leaky” vasculature), antibodies with little risk for immunogenicity, toxins with little risk for immunogenicity (e.g., protein toxins such as ricin would not be desired), and conjugation chemistry that allows for little off-target release of toxin, but where there is efficient release of toxin within target cells (i.e., in cases where this is required for efficacy). Successfully marketed antibodies include gemtuzumab ozogamicin, tositumomab, and ibritumomab tiuxetan. In each case, the antibodies target hematological cells. Tositumomab and ibritumomab utilize radioisotope toxins, where dissociation from the antibody is not required for the desired cytotoxic effect. Gemtuzumab ozogamicin employs a calicheamicin derivative toxin that is released in target cells after binding of gemtuzumab to the target receptor (CD33) and after receptor-mediated endocytosis of the immunotoxin. The toxin migrates to the nucleus and binds DNA, leading to double-strand breaks and cell death. There are few publications of PK/PD models for immunotoxin therapies. Ideally, mathematical models of immunotoxin pharmacokinetics and pharmacodynamics should account for the intact immunotoxin, “naked” antibody (i.e., antibody alone, following release of the toxin), and “free” toxin. In an interesting example, Zhu et al. applied physiologically based modeling and simulation to investigate relationships between the dose of radioimmunotoxins and uptake of the conjugates into tissue.50 Their modeling led to the conclusion that Fab fragments would be preferred for use in detection of tumors, whereas Fab2 fragments were predicted to be more effective for use in radioimmunotherapy.50 The structure of their PBPK model may be easily adapted to the prediction and characterization of the PK/PD of additional immunotoxins. Conclusions Antibody drugs demonstrate unique, complex PK characteristics. Absorption following IM or SC administration is slow and, for some antibodies, dose dependent. Antibody distribution kinetics is influenced by rates of convective transport, binding to tissue sites, and rates of catabolism within tissue. Traditional noncompartmental analyses and mammillary models may underestimate the steady-state distribution volume of many antibodies, particularly those associated with substantial elimination from tissue sites. Antibodies often demonstrate target-meditated disposition, VOLUME 84 NUMBER 5 | NOVEMBER 2008 | www.nature.com/cpt state where antibody–antigen binding influences the rate and extent of antibody distribution and elimination. FcRn, the Brambell receptor, protects IgG antibodies from elimination, but this protection system is saturable. Because of possible capacity limitations in presystemic catabolism, antibody binding to the target antigen, and FcRn-mediated transport, most antibodies demonstrate nonlinear, dose-dependent pharmacokinetics. Antibodies may be used for a variety of therapeutic applications, and several mechanisms may be associated with antibody effects. State-of-the-art mathematical models have, in many cases, allowed successful characterization of the plasma pharmacokinetics of antibody drugs and the time course of antibody effects. This research field is in its infancy, however, and there is great need to incorporate findings from basic research within mechanistic PK/PD models to facilitate efforts to predict antibody safety and efficacy in humans. Acknowledgments This work was supported by the University at Buffalo/Pfizer Strategic Alliance and by the National Institutes of Health grants HL67347, AI60687, and CA118213. Conflict of Interest The authors declared no conflict of interest. © 2008 American Society for Clinical Pharmacology and Therapeutics 1. Frazer, J.K. & Capra, J.D. Immunoglobulins: structure and function. In Fundamental Immunology 4th edn. (ed. Paul, W.E.) 37–74 (Lippincott-Raven, Philadelphia, PA, 1999). 2. Köhler, G. & Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495–497 (1975). 3. Lobo, E.D., Hansen, R.J. & Balthasar, J.P. Antibody pharmacokinetics and pharmacodynamics. J. Pharm. Sci. 93, 2645–2668 (2004). 4. Mould, D.R. & Sweeney, K.R. The pharmacokinetics and pharmacodynamics of monoclonal antibodies—mechanistic modeling applied to drug development. Curr. Opin. Drug Discov. Devel. 10, 84–96 (2007). 5. Supersaxo, A., Hein, W.R. & Steffen, H. Effect of molecular weight on the lymphatic absorption of water-soluble compounds following subcutaneous administration. Pharm. Res. 7, 167–169 (1990). 6. Kagan, L., Gershkovich, P., Mendelman, A., Amsili, S., Ezov, N. & Hoffman, A. The role of the lymphatic system in subcutaneous absorption of macromolecules in the rat model. Eur. J. Pharm. Biopharm. 67, 759–765 (2007). 7. Mortensen, D.L. et al. Pharmacokinetics and pharmacodynamics of multiple weekly subcutaneous efalizumab doses in patients with plaque psoriasis. J. Clin. Pharmacol. 45, 286–298 (2005). 8. Charman, S.A., Segrave, A.M., Edwards, G.A. & Porter, C.J. Systemic availability and lymphatic transport of human growth hormone administered by subcutaneous injection. J. Pharm. Sci. 89, 168–177 (2000). 9. Bitonti, A.J. et al. Pulmonary delivery of an erythropoietin Fc fusion protein in non-human primates through an immunoglobulin transport pathway. Proc. Natl. Acad. Sci. USA 101, 9763–9768 (2004). 10. Baxter, L.T., Zhu, H., Mackensen, D.G. & Jain, R.K. Physiologically based pharmacokinetic model for specific and nonspecific monoclonal antibodies and fragments in normal tissues and human tumor xenografts in nude mice. Cancer Res. 54, 1517–1528 (1994). 11. Ferl, G.Z., Wu, A.M. & DiStefano, J.J. 3rd. A predictive model of therapeutic monoclonal antibody dynamics and regulation by the neonatal Fc receptor (FcRn). Ann. Biomed. Eng. 33, 1640–1652 (2005). 12. Garg, A. & Balthasar, J.P. Physiologically-based pharmacokinetic (PBPK) model to predict IgG tissue kinetics in wild-type and FcRn-knockout mice. J. Pharmacokinet. Pharmacodyn. 34, 687–709 (2007). 13. Deane, R. et al. IgG-assisted age-dependent clearance of Alzheimer’s amyloid beta peptide by the blood-brain barrier neonatal Fc receptor. J. Neurosci. 25, 11495–11503 (2005). 14. Weinstein, J.N. et al. The pharmacology of monoclonal antibodies. Ann. NY Acad. Sci. 507, 199–210 (1987). 15. Mordenti, J. & Rescigno, A. Estimation of permanence time, exit time, dilution factor, and steady-state volume of distribution. Pharm. Res. 9, 17–25 (1992). art 16. Ng, C.M., Stefanich, E., Anand, B.S., Fielder, P.J. & Vaickus, L. Pharmacokinetics/ pharmacodynamics of nondepleting anti-CD4 monoclonal antibody (TRX1) in healthy human volunteers. Pharm. Res. 23, 95–103 (2006). 17. Hayashi, N., Tsukamoto, Y., Sallas, W.M. & Lowe, P.J. A mechanism-based binding model for the population pharmacokinetics and pharmacodynamics of omalizumab. Br. J. Clin. Pharmacol. 63, 548–561 (2007). 18. Lammerts van Bueren, J.J. et al. Effect of target dynamics on pharmacokinetics of a novel therapeutic antibody against the epidermal growth factor receptor: implications for the mechanisms of action. Cancer Res. 66, 7630–7638 (2006). 19. Brambell, F.W., Hemmings, W.A. & Morris, I.G. A theoretical model of gammaglobulin catabolism. Nature 203, 1352–1354 (1964). 20. Junghans, R.P. Finally! The Brambell receptor (FcRB). Mediator of transmission of immunity and protection from catabolism for IgG. Immunol. Res. 16, 29–57 (1997). 21. Waldmann, T.A. & Strober, W. Metabolism of immunoglobulins. Prog. Allergy 13, 1–110 (1969). 22. Ober, R.J., Radu, C.G., Ghetie, V. & Ward, E.S. Differences in promiscuity for antibody-FcRn interactions across species: implications for therapeutic antibodies. Int. Immunol. 13, 1551–1559 (2001). 23. Jin, F. & Balthasar, J.P. Mechanisms of intravenous immunoglobulin action in immune thrombocytopenic purpura. Hum. Immunol. 66, 403–410 (2005). 24. Getman, K.E. & Balthasar, J.P. Pharmacokinetic effects of 4C9, an anti-FcRn antibody, in rats: implications for the use of FcRn inhibitors for the treatment of humoral autoimmune and alloimmune conditions. J. Pharm. Sci. 94, 718–729 (2005). 25. Mezo, A.R., McDonnell, K.A., Castro, A. & Fraley, C. Structure-activity relationships of a peptide inhibitor of the human FcRn:human IgG interaction. Bioorg. Med. Chem. 16, 6394–6405 (2008). 26. Hwang, W.Y. & Foote, J. Immunogenicity of engineered antibodies. Methods 36, 3–10 (2005). 27. Hakimi, J. et al. Reduced immunogenicity and improved pharmacokinetics of humanized anti-Tac in cynomolgus monkeys. J. Immunol. 147, 1352–1359 (1991). 28. Kimball, J.A. et al. The OKT3 Antibody Response Study: a multicentre study of human anti-mouse antibody (HAMA) production following OKT3 use in solid organ transplantation. Transpl. Immunol. 3, 212–221 (1995). 29. Svenson, M., Geborek, P., Saxne, T. & Bendtzen, K. Monitoring patients treated with anti-TNF-alpha biopharmaceuticals: assessing serum infliximab and antiinfliximab antibodies. Rheumatology (Oxford) 46, 1828–1834 (2007). 30. Stephens, S. et al. Comprehensive pharmacokinetics of a humanized antibody and analysis of residual anti-idiotypic responses. Immunology 85, 668–674 (1995). 31. Schellekens, H. Immunogenicity of therapeutic proteins: clinical implications and future prospects. Clin. Ther. 24, 1720–1740; discussion 1719 (2002). 32. Rehlaender, B.N. & Cho, M.J. Antibodies as carrier proteins. Pharm. Res. 15, 1652–1656 (1998). 33. Mordenti, J., Chen, S.A., Moore, J.A., Ferraiolo, B.L. & Green, J.D. Interspecies scaling of clearance and volume of distribution data for five therapeutic proteins. Pharm. Res. 8, 1351–1359 (1991). 34. Grene-Lerouge, N.A., Bazin-Redureau, M.I., Debray, M. & Scherrmann, J.M. Interspecies scaling of clearance and volume of distribution for digoxinspecific Fab. Toxicol. Appl. Pharmacol. 138, 84–89 (1996). 35. Woo, S. & Jusko, W.J. Interspecies comparisons of pharmacokinetics and pharmacodynamics of recombinant human erythropoietin. Drug Metab. Dispos. 35, 1672–1678 (2007). 36. Vugmeyster, Y., Szklut, P., Tchistiakova, L., Abraham, W., Kasaian, M. & Xu, X. Preclinical pharmacokinetics, interspecies scaling, and tissue distribution of humanized monoclonal anti-IL-13 antibodies with different IL-13 neutralization mechanisms. Int. Immunopharmacol. 8, 477–483 (2008). 37. Duconge, J., Fernandez-Sanchez, E. & Alvarez, D. Interspecies scaling of the monoclonal anti-EGF receptor ior EGF/r3 antibody disposition using allometric paradigm: is it really suitable? Biopharm. Drug. Dispos. 25, 177–186 (2004). 38. Furuya, Y., Ozeki, T., Takayanagi, R., Yokoyama, H., Okuyama, K. & Yamada, Y. Theory based analysis of anti-inflammatory effect of infliximab on Crohn’s disease. Drug Metab. Pharmacokinet. 22, 20–25 (2007). 39. Meno-Tetang, G.M. & Lowe, P.J. On the prediction of the human response: a recycled mechanistic pharmacokinetic/pharmacodynamic approach. Basic Clin. Pharmacol. Toxicol. 96, 182–192 (2005). 40. Marathe, A., Peterson, M.C. & Mager, D.E. Integrated cellular bone homeostasis model for denosumab pharmacodynamics in multiple myeloma patients. J. Pharmacol. Exp. Ther. 326, 555–562 (2008). 41. Cartron, G. et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 99, 754–758 (2002). Clinical pharmacology & Therapeutics | VOLUME 84 NUMBER 5 | NOVEMBER 2008 557 state art 42. Meijer, R.T., Koopmans, R.P., ten Berge, I.J. & Schellekens, P.T. Pharmacokinetics of murine anti-human CD3 antibodies in man are determined by the disappearance of target antigen. J. Pharmacol. Exp. Ther. 300, 346–353 (2002). 43. Mould, D.R. et al. A population pharmacokinetic-pharmacodynamic analysis of single doses of clenoliximab in patients with rheumatoid arthritis. Clin. Pharmacol. Ther. 66, 246–257 (1999). 44. Sharma, A. et al. Comparative pharmacodynamics of keliximab and clenoliximab in transgenic mice bearing human CD4. J. Pharmacol. Exp. Ther. 293, 33–41 (2000). 45. Deng, R. & Balthasar, J.P. Pharmacokinetic/pharmacodynamic modeling of IVIG effects in a murine model of immune thrombocytopenia. J. Pharm. Sci. 96, 1625–1637 (2007). 46. Mager, D.E., Mascelli, M.A., Kleiman, N.S., Fitzgerald, D.J. & Abernethy, D.R. Simultaneous modeling of abciximab plasma concentrations and ex 558 47. 48. 49. 50. vivo pharmacodynamics in patients undergoing coronary angioplasty. J. Pharmacol. Exp. Ther. 307, 969–976 (2003). Bauer, R.J., Dedrick, R.L., White, M.L., Murray, M.J. & Garovoy, M.R. Population pharmacokinetics and pharmacodynamics of the anti-CD11a antibody hu1124 in human subjects with psoriasis. J. Pharmacokinet. Biopharm. 27, 397–420 (1999). Ng, C.M., Joshi, A., Dedrick, R.L., Garovoy, M.R. & Bauer, R.J. Pharmacokineticpharmacodynamic-efficacy analysis of efalizumab in patients with moderate to severe psoriasis. Pharm. Res. 22, 1088–1100 (2005). Luo, F.R. et al. Correlation of pharmacokinetics with the antitumor activity of Cetuximab in nude mice bearing the GEO human colon carcinoma xenograft. Cancer Chemother. Pharmacol. 56, 455–464 (2005). Zhu, H., Baxter, L.T. & Jain, R.K. Potential and limitations of radioimmunodetection and radioimmunotherapy with monoclonal antibodies. J. Nucl. Med. 38, 731–741 (1997). VOLUME 84 NUMBER 5 | NOVEMBER 2008 | www.nature.com/cpt