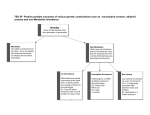
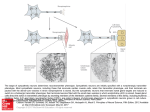
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Official Journal of ESPE European Society for Paediatric Endocrinology SLEP Sociedade Latino-Americana de Endocrinologia Pediátrica HORMONE RESEARCH From Developmental Endocrinology to Clinical Research Asia Pacific Pediatric Endocrine Society Founded 1970 as ‘Hormones’ by M. Marois Continued by J. Girard (1976–1995); M.B. Ranke (1996–2003) Editor-in-Chief Editorial Board P. Czernichow, Paris I.J.P. Arnhold, São Paulo W.F. Blum, Bad Homburg J.-P. Bourguignon, Liège F. Cassorla, Santiago J.-P. Chanoine, Vancouver M. Dattani, London H. Delemarre-van de Waal, Amsterdam M. Donaldson, Glasgow D.B. Dunger, Cambridge L. Dunkel, Kuopio U. Feldt-Rasmussen, Copenhagen M.G. Forest, Lyon K. Fujieda, Asahikawa Associate Editors J.-C. Carel, Paris P.E. Clayton, Manchester W.S. Cutfield, Auckland O. Hiort, Lübeck P.E. Mullis, Bern M.A. Rivarola, Buenos Aires P. Saenger, Bronx, N.Y. M.O. Savage, London O. Söder, Stockholm G. Van Vliet, Montreal J.J. Heinrich, Buenos Aires A.C. Hokken-Koelega, Rotterdam A. Hübner, Dresden L. Ibañez, Barcelona K.Y. Loke, Singapore C.J. Migeon, Baltimore, Md. J.P. Monson, London M. Phillip, Petah Tiqwa M. Polak, Paris M.B. Ranke, Tübingen R.G. Rosenfeld, Palo Alto, Calif. S.M. Shalet, Manchester T. Tanaka, Tokyo R.J. Voutilainen, Kuopio ESPE Council 2006–2007 President R. Voutilainen, Kuopio President-Elect Atilla Büyükgebiz, Istanbul Secretary General F. Chiarelli, Chieti Chairman of Finance Committee O. Söder, Stockholm Chairman of Clinical Practice Committee S. Cianfarani, Rome Chairman of Education and Training Committee S. Drop, Rotterdam Chairman of Programme Organising Committee Z. Hochberg, Haifa Chairman of Corporate Liaison Board C.J.H. Kelnar, Edinburgh Member J. Léger, Paris Printed in Switzerland on acid-free and non-aging paper (ISO 9706) by Reinhardt Druck, Basel Appears monthly: 2 volumes per year (12 issues) S. Karger Medical and Scientific Publishers Basel • Freiburg • Paris • London New York • Bangalore • Bangkok Singapore • Tokyo • Sydney Disclaimer The statements, options and data contained in this publication are solely those of the individual authors and contributors and not of the publisher and the editor(s). The appearance of advertisements in the journal is not a warranty, endorsement, or approval of the products or services advertised or of their effectiveness, quality or safety. The publisher and the editor(s) disclaim responsibility for any injury to persons or property resulting from any ideas, methods, instructions or products referred to in the content or advertisements. Drug Dosage The authors and the publisher have exerted every effort to ensure that drug selection and dosage set forth in this text are in accord with current recommendations and practice at the time of publication. However, in view of ongoing research, changes in government regulations, and the constant flow of information relating to drug therapy and drug reactions, the reader is urged to check the package insert for each drug for any change in indications and dosage and for added warnings and precautions. This is particularly important when the recommended agent is a new and/or infrequently employed drug. Fax +41 61 306 12 34 E-Mail [email protected] www.karger.com All rights reserved. No part of this publication may be translated into other languages, reproduced or utilized in any form or by any means, electronic or mechanical, including photocopying, recording, microcopying, or by any information storage and retrieval system, without permission in writing from the publisher or, in the case of photocopying, direct payment of a specified fee to the Copyright Clearance Center (see ‘General Information’). © Copyright 2007 by J.M. Wit, M.B. Ranke, C.J.H. Kelnar Published by S. Karger AG P.O. Box, CH–4009 Basel (Switzerland) Printed in Switzerland on acid-free and non-aging paper (ISO 9706) by Reinhardt Druck, Basel ISBN 978–3–8055–8334–3 ESPE Classi f icat ion of Pae diat ric E ndoc rine Diagnoses Edited by Jan M. Wit Michael B. Ranke Christopher J.H. Kelnar With contributions by Hans Akerblom Albert Aynsley-Green Giampiero Baroncelli Jean-Pierre Bourguignon Henriette A. Delemarre-Van de Waal Malcolm Donaldson Herwig Frisch Annette Grüters-Kieslich Lars Hagenäs Eberhard Heinze Ieuan A. Hughes Daniel Iliev Brygida Koehler Klaus Kruse Agne Larsson Jörn Müller Stefan Riedl Martin Ritzén Tomas E. Romer Werner Rosendahl Giuseppe Saggese Niels Skakkebaek Yara Smit Gyula Soltész J. M.W. was supported by a grant of the Sabbatical Leave Programme of the European Society for Paediatric Endocrinology, supported by Eli Lilly & Co. CO NTE NTS Foreword . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . VII User Guide. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . IX 1 SHORT STATURE 1A 1B 1C 2 Precocious Puberty . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Single Variations of Normal Puberty . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Contrasexual Pubertal Development . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Delayed Puberty . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Menstrual Disorders . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14 15 15 16 16 DISORDERS OF SEX DE VELOPMENT DSD 4A 4B 4C 4D 5 Primary Growth Disorders . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10 Secondary Growth Disorders . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11 Idiopathic (Normal Variant Tall Stature) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12 DISORDERS OF PUBERT Y 3A 3B 3C 3D 3E 4 1 2 5 TALL STATURE 2A 2B 2C 3 Primary Growth Failure. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Secondary Growth Failure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Idiopathic Short Stature. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sex Chromosome Disorders of Sex Development . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 46,XY Disorders of Sex Development . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 46,XX Disorders of Sex Development . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Unclassified Forms of Abnormal Sexual Development/Anatomical Disruptions. . 21 22 23 24 OVERWEIGHT AND OBESIT Y 5A 5B 5C 5D 5E 5F ‘Simple’ Obesity, Common Obesity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Genetic Causes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Endocrine Disorders . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Central Nervous System Causes (Central Obesity) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Obesity Accompanying Immobility, Mental Disturbances, Social and Cultural Pressure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Iatrogenic Obesity due to Medication. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ESPE Classification of Paediatric Endocrine Diagnoses 27 27 28 28 28 29 IV 6 PITUITARY, HYPOTHAL AMUS, CENTR AL NERVOUS SYSTEM CNS Section 1: Functional Classification. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6A Deficiencies of Anterior Pituitary Hormones . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6B Overproduction of Anterior Pituitary Hormones . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6C Central Diabetes Insipidus. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6D Hypothalamic Dysfunction, Not Classified Elsewhere. . . . . . . . . . . . . . . . . . . . . . . . . . . 31 31 33 33 33 Section 2: Aetiological Classification . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 33 6E Congenital Disorders. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 33 6F Acquired Disorders . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34 7 THYROID DISORDERS 7A 7B 7C 7D 7E 8 Primary Adrenal Insufficiency . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Secondary Adrenal Insufficiency. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Adrenal Excess. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disorders of the Adrenal Medulla . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 55 56 56 57 TESTICUL AR DISORDERS/DISORDERS OF MALE GENITALS 9A 9B 9C 9D 9E 9F 9G 9H 9Y 9Z 10 44 45 46 46 47 ADRENAL DISORDERS 8A 8B 8C 8D 9 Hypothyroidism . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Hyperthyroidism. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Goitre . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Thyroid Tumours. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Other Thyroid Disorders. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Hypergonadotrophic Hypogonadism. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Cryptorchidism/Maldescended Testes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Acquired Testicular Disorders. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Tumours of Testes. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disorders of Penis. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Scrotal Disorders. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disorders of the Epididymis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disorders of Testicular Blood Vessels . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Other Specified Disorders of the Male Genitalia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Other Disorders of the Male Genitalia, Unspecified . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 63 64 64 65 65 66 66 66 66 66 OVARIES, FEMALE REPRODUC TIVE TR AC T AND BREASTS 10A 10B 10C 10D Ovary . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disorders of the Uterus and Cervix. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disorders of the Vagina and External Female Genitalia . . . . . . . . . . . . . . . . . . . . . . . . . Disorders of the Breast . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 68 69 70 71 V 11 DISORDERS OF GLUCOSE AND TRIGLYCERIDE ME TABOLISM 11A Diabetes Mellitus . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 73 11B Hypoglycaemia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 75 11C Primary Disturbances of Lipoprotein Metabolism . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 77 12 DISORDERS OF BONE METABOLISM, INCLUDING C ALCIUM/ PHOSPHATE ME TABOLISM 12A 12B 12C 12D 12E 13 Transient Hypocalcaemia (Neonatal). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Permanent Hypocalcaemia. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Rickets . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Osteoporosis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Hypercalcaemia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 87 87 89 90 90 DISORDERS OF WATER BAL ANCE 13A Disorders Characterized by Polydipsia and Polyuria . . . . . . . . . . . . . . . . . . . . . . . . . . . . 96 13B Disorders Characterized by Hypernatraemia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 97 13C Disorders Characterized by Hyponatraemia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 97 14 SYNDROMES WITH ENDOCRINE FEATURES 14A Chromosomal Abnormalities . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 101 14B Congenital Dysmorphic Syndromes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 102 14C Non-Dysmorphic Syndromes. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 106 List of Abbreviations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 120 ESPE Classification of Paediatric Endocrine Diagnoses VI FO R EWO R D The ESPE Classification of Paediatric Endocrine Diagnoses One of the cornerstones of medical practice is getting the diagnosis correct. After a diagnosis is made, clinicians usually like to store the information about the diagnoses of individual patients in their files and registries. This can then improve patient care, because it makes information about the natural history and treatment responses available of patients that had been seen in the past. It can also be helpful for scientific research, for example in collecting data on patient groups. Until now, no list of paediatric endocrine diagnoses has been available. Current diagnostic classifications for the whole field of medicine lack the detail that is requested by the subspecialist. In some countries, a specific diagnostic classification for paediatrics is available, but even that lacks the necessary detail. This was the main reason which prompted us to produce the ESPE Classification of Paediatric Endocrine Diagnoses that forms the body of this publication. We believe that a classification of diagnoses is an important tool in achieving optimal communication between paediatric endocrinologists, as it enables physicians around the world to speak the same language. In designing a formal coding system, the following requirements should be considered: (1) it should allow classification of all patients in question; (2) there should be room for accommodating new aetiologies and pathogenetic aspects; (3) the diagnoses listed should be well defined, making misclassifications virtually impossible; (4) the classifications should follow one general principle (e.g. nosology, aetiology, pathogenesis or symptomatology); (5) it should be easy to use, and (6) it should optimally serve the purpose for which it was designed. As each diagnosis can be classified according to different aspects, for example function or anatomy, our choices are to some extent subjective. However, we have tried to follow the logic of the paediatric endocrine clinician as much as possible, so that it would be as easy as possible to find the diagnosis in the structure of each chapter. We have extensively made use of the lists of causes of disorders as presented in various (paediatric) endocrine textbooks. This list of preconditions makes it obvious that no system for the classification of diseases can be perfect. The authors have attempted to develop a system of diagnoses in paediatric endocrinology that serves both scientific and practical requirements. In addition to establishing a coding system, a glossary of the most important disorders has been appended, in the form of endnotes, for the purpose of clarification and unequivocal description. Nevertheless, in areas where we are still ignorant about aetiology, any classification will be, to an extent, arbitrary (for example ‘idiopathic short/tall stature’, ‘familial’, ‘normal variant’). The reader should, therefore, keep in mind that in some situations a classification which is still necessarily descriptive can, if used uncritically, obscure rather than il- VII luminate understanding. In addition, when thinking about biological systems, “there is no law of nature which says boundaries have to be clear cut” [1]. Keeping in mind, therefore, the fact that our understanding of endocrinological diseases is developing rapidly, we have left room for additions and modifications in this classification system. We have added the codes for the ICD-10 classification, so that the ESPE Classification can be relatively easily incorporated into a hospital registry. In addition, where applicable, OMIM codes are given for further and updated information about the various syndromes that have endocrine elements. The editors thank all the collaborators for their valuable contributions and hope that this classification will serve the paediatric endocrine community well. J.M. Wit, M.B. Ranke, C.J.H. Kelnar March 2007 Reference 1 Dawkins R: Gaps in the Mind; in: A Devil’s Chaplain – Selected Essays. London, Weidenfeld & Nicolson, 2003. ESPE Classification of Paediatric Endocrine Diagnoses VIII USE R GU I D E In the preparation of this classification we have used several strategies and rules. In the following paragraphs these are explained for the purpose of easy use. 1 The diagnostic classification is built as a tree structure, with generally up to 5 levels (rarely going up to 6 levels). We have chosen a system of LETTERS-numbers-letters-numbers-letters(-numbers), which is easier to use than a system only consisting of numbers. The first number used is 1, except for the box with ‘Disorders classified elsewhere’, which gets a ‘0’. The first letter is ‘a’, also for boxes with ‘Disorders classified elsewhere’. 2 Each diagnosis has a main site (indicated as [primary]) and can have one or more secondary sites (indicated as [secondary]). Additional information, including the footnotes, ICD-10 codes, and OMIM codes, is only given at the primary site of classification. If the disorder is also coded in other chapters, or other segments of the same chapter, this is mentioned at the beginning of the pertinent section, under the heading: ‘Disorders classified elsewhere’ (coded as ‘0’ or ‘a’). The diagnoses that are classified elsewhere are indicated in italics. 3 Where applicable, it is indicated which diagnosis is not included in the pertinent section. This is written as: ‘Excluded:’, followed by the diagnoses that are excluded. 4 Where applicable, the lists of diagnoses under a certain group end with a section called ‘other specified disorders’ (labelled as 8, 88, or y) and a section called ‘other disorders, unspecified’ (labelled as 9, 99, or z). These serve to code conditions that cannot be found in the current classification. 5 With respect to spelling, we have followed the British English spelling. We have used no ’s to add to the names of the discoverers of the ‘syndromes’ (e.g. Turner syndrome instead of Turner’s syndrome), but the ’s has been used for the names of ‘Diseases’ (e.g. Addison’s disease). 6 Suggestions for coding are printed in italics. 7 Explanatory texts are given in the form of footnotes, and have been limited to 3 short sections: synonyms; phenotype, including concise information about symptoms, signs, laboratory investigations, radiology, and other further investigations, and comments. In some instances a reference is added. 8 Hormonal findings are indicated in an abbreviated form. First, hormones are abbreviated in their usual fashion (see list of abbreviations). Second, ‘increased’ is indicated as ‘+’, ‘decreased’ as ‘–’, and ‘normal’ as ‘N’, printed immediately behind the abbreviated hormone (e.g. T (+) means increased testosterone concentration). 9 If a child has a disorder that expresses itself in different endocrine systems, we advise to classify the condition according to various systems. This also applies to the codes given to ‘Classified elsewhere’. So, the short, obese, hypogonadic and diabetic child with Prader-Willi-Labhart syndrome may be coded 14B.25 (primary), 1A.1a (short), 5B.2a (obesity), 6A.3d (hypogonadotrophic hypogonadism) and 11A.3h.10 (diabetes mellitus). J.M. Wit, M.B. Ranke, C.J.H. Kelnar IX ESPE Code Diagnosis 1 S H O R T STATU R E 1 1A PR I M ARY G R OW T H FAI LU R E 1A.1 Clinically defined syndromes Syndromes classified elsewhere: 45,X/46,XY disorder of sex development (4A.1) 45,X and variants with female phenotype ((Ulrich)-Turner syndrome) (14A.5) Phenotypic male with X/XY mosaicism (14A.4) 18q deletion syndrome (14A.1) Aarskog-Scott syndrome (14B.1) Bloom syndrome (14B.6) Cornelia de Lange syndrome (14B.9) DiGeorge syndrome (velocardiofacial syndrome) (14B.10) Down syndrome (14A.2) Kabuki makeup syndrome (14C.4f) Noonan syndrome (14B.24) Prader-Willi-Labhart syndrome (14B.25) Von Recklinghausen’s disease (neurofibromatosis type 1) (14B.27) Rubinstein-Taybi syndrome (14B.29) Silver-Russell syndrome (14B.31) Williams-Beuren syndrome (14B.37) Other specified syndromes, e.g. Three M slender-boned dwarfism, Seckel syndrome, Mulibrey nanism Other syndromes associated with short stature, unspecified 1A.1a 1A.1y 1A.1z 1A.2 1A.2a 1A.2y 1A.2z 1A.3 1A.3a 1A.3a.1 1A.3a.2 OMIM ICD-10 Small for gestational age (SGA) with failure of catch-up growth2 Disorders classified elsewhere: IGF-I deficiency (1B.4e) IGF resistance (1B.4f) Due to known cause (specify), e.g. prenatal infection, drugs, smoking, alcohol Idiopathic P05.1 Skeletal dysplasias (constitutional disorders of bone)3 Achondroplasia group (group 1)4 Achondroplasia Hypochondroplasia Q77, Q78 Short Stature P00.2–4 #100800 #146000 1 ESPE Code Diagnosis 1A.3a.8 Other specified disorders included in this group (thanatophoric dysplasia, SADDAN) Type II collagenopathies (COL2A1 defects) (group 8) Spondyloepiphyseal dysplasia congenita Other specified disorders in this group Mesomelic dysplasias (group 16) Dyschondrosteosis (Leri-Weill and other defects in the SHOX gene, e.g. in children without dysmorphic features) [primary: 14B.19] Langer type (homozygous dyschondrosteosis) Other specified disorders included in this group Dysostosis multiplex group (group 22) Mucopolysaccharidosis (type IH, IS, II–VII) Mucolipidosis (type II and III) 1A.3b 1A.3b.1 1A.3b.8 1A.3c 1A.3c.1 1A.3c.2 1A.3c.8 1A.3d 1A.3d.1 1A.3d.2 OMIM ICD-10 #183900 #127300 #249700 E76 #252500 E77.0 #252600 1A.3z Other specified disorders included in this group Dysplasias with decreased bone density (group 24) Osteogenesis imperfecta I–VI [primary: 12D.1b] Other specified disorders included in this group Dysplasias with defective mineralisation (group 25) Hypophosphatasia Hypophosphataemic rickets [primary 12C.2] Other specified disorders included in this group Disorders included in other groups (2–7, 9–15, 17–21, 23, 26–33) Other skeletal dysplasia, unspecified 1B S E CO N DARY G R OW T H FAI LU R E 1B.1 Insufficient nutrient intake (malnutrition) E40–46 1B.2 Disorders in organ systems Cardiac disorders Pulmonary disorders, e.g. cystic fibrosis Q20–28 J40–99 1A.3d.8 1A.3e 1A.3e.1 1A.3e.8 1A.3f 1A.3f.1 1A.3f.2 1A.3f.8 1A.3y 1B.2a 1B.2b #241500 #307800 (E84) 1B.2c 1B.2d 1B.2e Liver disorders Intestinal disorders, e.g. Crohn’s disease, malabsorption syndromes, short bowel syndrome Renal disorders, e.g. Fanconi syndrome, renal acidosis K70–77 K50–52 K90–93 N10–19 N25–29 1B.2f 1B.2g Chronic anaemia Multiorgan disorders ESPE Classification of Paediatric Endocrine Diagnoses D50–64 2 ESPE Code Diagnosis OMIM 1B.2h Muscular and neurological disorders, e.g. Duchenne muscular dystrophy, congenital myotonia Connective tissue diseases, e.g. juvenile arthritis Other specified organ or systemic disorders G71–73 1B.2i 1B.2y 1B.3 1B.3a 1B.3a.0 1B.3a.1 1B.3a.2 1B.3a.2a 1B.3a.2b 1B.3a.2c 1B.3a.2d 1B.3a.2e 1B.3a.2f 1B.3a.2g 1B.3a.2y 1B.3a.3 1B.3a.4 1B.3a.8 1B.3a.9 1B.3a.9a 1B.3a.9b Growth hormone deficiency (secondary IGF deficiency)5 In case of multiple pituitary deficiencies, classify the various deficiencies separately: ACTH deficiency (6A.1) TSH deficiency (6A.2) Gonadotropin deficiency (6A.3) Prolactin deficiency (6A.4) Vasopressin deficiency (diabetes insipidus) (13A.1) Congenital growth hormone deficiency Disorders classified elsewhere: Associated with complex syndromes: Fanconi renotubular syndrome (14B.13) Rieger syndrome (14B.28) Kabuki make-up syndrome (14C.4f) Associated with other complex syndromes: ectodactyly-ectodermal dysplasia clefting syndrome Known genetic defects – HESX1 – LHX3 – LHX4 – PROP1 – POU1F1 – GHRHR – GH – Other specified genetic defects – Associated with cerebral or facial malformations, e.g. septooptic dysplasia [primary 6E.1a], empty sella syndrome, solitary central maxillary incisor syndrome, mid-line palateal cleft, arachnoid cyst, congenital hydrocephalus, hypoplastic anterior pituitary + missing stalk + ectopic posterior pituitary (HME) Excluded: Known genetic defects (1B.3a.2) – Associated with prenatal infection, e.g. rubella – Associated with other specified disorders – Idiopathic – ‘Classical’ idiopathic growth hormone deficiency6 – Neurosecretory dysfunction7 Short Stature ICD-10 M08 E23.0 E23.0 E23.0 E23.0 *601802 *600577 *602146 *601538 +173110 *139191 +139250 Q04.4 Q37.9 O35.0 P35.0 E23.0 E23.0 3 ESPE Code Diagnosis 1B.3b Acquired growth hormone deficiency – Craniopharyngioma – Other pituitary tumours, e.g. germinoma, hamartoma – Cranial tumours distant from the pituitary/hypothalamic area, e.g. astrocytoma, ependymoma, glioma, medulloblastoma, nasopharyngeal tumour 1B.3b.1 1B.3b.2 1B.3b.3 OMIM ICD-10 E23.0 D44.4 M9064/3 C71 M9400/3 M9391/3 M9380/3 M9470/3 IB.3b.4 IB.3b.5 IB.3b.6 IB.3b.7 IB.3b.8 IB.3b.9 1B.4 1B.4a 1B.4b 1B.4c 1B.4d 1B.4e 1B.4f 1B.4z 1B.5 1B.5a 1B.5b 1B.5y 1B.6 1B.6a 1B.6b 1B.6c – Tumours outside the cranium, e.g. leukaemia, lymphoma – Head trauma – Central nervous system infection – Granulomatous diseases, e.g. histiocytosis – Vascular anomaly – Other causes, unspecified Other disorders of the growth hormone-IGF axis (primary IGF-I deficiency and resistance) Bio-inactive growth hormone (Kowarski syndrome)8 Abnormalities of the growth hormone receptor (growth hormone insensitivity syndrome, Laron syndrome) Abnormalities of GH signal transduction, e.g. STAT5B defect ALS (acid-labile subunit) deficiency IGF-I deficiency IGF resistance (IGF1R defects, postreceptor defects) Other disorders, unspecified Other endocrine disorders Disorders classified elsewhere: Cushing syndrome (8C.1) Hypothyroidism (7A) Leprechaunism (11A.3b.2) Poorly controlled diabetes mellitus, Mauriac syndrome (14C.2) Short adult stature caused by accelerated bone maturation, e.g. precocious puberty (3A), hyperthyroidism (7B), congenital adrenal hyperplasia (8A.1), exogenous estrogens or androgens (3A.2d) Other specified disorders Metabolic disorders Disorders classified elsewhere: Disorders of calcium and phosphorus metabolism (1A.3 and 12) Disorders of carbohydrate metabolism Disorders of lipid metabolism ESPE Classification of Paediatric Endocrine Diagnoses C91–96 S06 G01–08 D76 Q28 E23.0 #262650 E34.3 #262500 E34.3 #245590 E34.3 +601489 E34.3 #608747 E34.3 #270450 E34.3 E34.3 E74 E75 4 ESPE Code Diagnosis 1B.6d Disorders of protein metabolism Other specified metabolic disorders Other metabolic disorders, unspecified E70–72 Psychosocial Psychosocial (emotional) deprivation9 Anorexia nervosa Depression Other specified psychosocial causes E34.3 1B.6y 1B.6z 1B.7 1B.7a 1B.7b 1B.7c 1B.7y OMIM ICD-10 E76–83 E88 T74 F50 F32.9 E34.3 1B.8y Iatrogenic Systemic glucocorticoid therapy [primary 8C.1c] Local glucocorticoid therapy (inhalation, intestinal, other) Other medication Treatment of childhood malignancy Total body irradiation Chemotherapy Other specified iatrogenic causes 1C I D I O P AT H I C S H O R T S T AT U R E 10 E34.3 1C.1 Familial (idiopathic) short stature11 With normal pubertal onset With delayed pubertal onset Onset of puberty not yet known12 Onset of puberty unknown E34.3 Non-familial (idiopathic) short stature13 With normal pubertal onset With delayed pubertal onset (constitutional delay in growth and puberty, or constitutional delay in growth and adolescence) Onset of puberty not yet known14 Onset of puberty unknown E34.3 1B.8 1B.8a 1B.8b 1B.8c 1B.8d 1B.8d.1 1B.8d.2 1C.1a 1C.1b 1C.1c 1C.1d 1C.2 1C.2a 1C.2b 1C.2c 1C.2d Short Stature T38.0 T49.0 T36–50 T66 T45.1 T78.9 5 1 Short stature Comments: Short stature is defined as a height below –2.0 SDS (2.3rd percentile) for a given age, sex and population. It is the result of impaired bone growth in the foregoing period, which is expressed in terms of a reduced length/height velocity in at least some period of life (including the intrauterine period). In some conditions growth velocity is only diminished early in life (e.g. in children born SGA with failure of catch-up growth), in other conditions growth velocity is continuously low (e.g. in children with complete growth hormone deficiency). The terms ‘growth delay’ and ’growth retardation‘ are also frequently used with the same intention as short stature. In this diagnostic classification an abnormally low height velocity is also classified under the general heading of ’Short stature‘. The causes of short stature are varied and differ with respect to aetiology and pathogenesis. To our present knowledge, disorders of the hormonal system account only for a minority of children with short stature. Since growth disorders are the key symptom of many hormonal abnormalities and since children with growth disorders are commonly presented to the paediatric endocrinologist, disorders presenting with short/tall stature are part of this classification system. 2 Small for gestational age (SGA) with failure of catch-up growth Phenotype: Short stature and birth length and/or weight below –2 SDS (or below the 2.3rd centile) for gestational age for the reference population. Comments: There is some discussion about the use of the terms intrauterine growth retardation (IUGR) and small for gestational age (SGA). There appears now some consensus that the term IUGR implies impaired fetal growth and should be considered a prenatal diagnosis. SGA describes birth length and/or weight in relation to gestational age and is a post-partum diagnosis. The majority of babies born SGA (approximately 85%) catch up in height (within 2–3 years) and will not be short in childhood or in adulthood, although their mean height is somewhat lower than expected for target height. The 15% of babies who do not catch-up (height < –2 SDS after 2 or 3 years) are termed SGA with failure to catch-up, or shortly SGA. Those who failed to catch-up by 2 years are at high risk of remaining short in later life. Thus, the term SGA is purely descriptive, and is a working diagnosis for children born small and continuing to be short. In most cases there is no known aetiology and/or pathogenesis. 3 Skeletal dysplasias (constitutional disorders of bone) Comments: Skeletal dysplasias exist in a great variety and are commonly associated with short stature. Traditionally, the diagnosis was established based on the clinical appearance during development and conventional X-ray findings. More recently, the classification of skeletal dysplasias has been based upon the expanding understanding of the pathophysiology of bone development and the discovery of the underlying molecular genetic defects. This has resulted in an ’International nomenclature and classification of osteochondrodysplasias (1997)’ published by the International Working Group on Constitutional Diseases of Bone [Am J Med Genet 1998;79:376–382] and a more recent update [Hall CM: International nosology and classification of constitutional disorders of bone (2001). Am J Med Genet 2002;113:65–77]. In this classification 33 groups are presented, each including various clinical entities. For the ESPE Classification of Paediatric Endocrine Diagnoses just 6 groups out of the 33 are mentioned specifically. For a more detailed classification we refer to the most recent paper on International Nosology and Classification. ESPE Classification of Paediatric Endocrine Diagnoses 6 4 Achondroplasia, hypochondroplasia Phenotype: Achondroplasia and hypochondroplasia are disorders characterised by disproportionate short stature with rhizomelic limb shortening. They are commonly caused by mutations in the tyrosine kinase domain of the fibroblast growth factor receptor 3 (FGFR3) gene. However, in the absence of these mutations the diagnosis may still be confirmed by typical radiological findings. 5 Growth hormone deficiency (GHD) Synonym (if combined with other deficiencies): Hypothalamo-hypopituitarism, multiple congenital pituitary hormone deficiency. Phenotype: Growth retardation, retarded bone age, hypoglycaemia, truncal obesity, symptoms of neonatal hypoglycaemia and those of associated anomalies, e.g. midline defects, micropenis. Comments: An impaired secretion of GH in children is causally associated with impaired growth, changes of the anthropometrical characteristics (e.g. normal proportions, relatively large head) and the composition of the body (e.g. sarcopenia, excess of body fat) as well as functional abnormalities (e.g. hypoglycaemia). Classifications based on functional investigations of GH secretion and action have allowed some degree of understanding of the role of the various components of the GH-IGF-I axis. Functional investigations include measurements of GH by means of various assays (immunoassays, bioassays), determination of GH levels after various exogenous stimuli, the analysis of spontaneously secreted GH, measurements of GH dependent factors (IGF-I, IGFBP-3; ALS), and the biochemical response to exogenous GH (e.g. IGF generation). In addition to the difficulty of conducting such functional tests, the limitations of standardisation and the lack of normative data has hindered a concise interpretation of the obtained data and their interplay until today. New imaging tools (CT, MRT) allow the description of qualitative and quantitative anatomical anomalies of the pituitary region. A higher resolution and functional imaging is likely to lead to a further dimension in describing more subtle abnormalities. Discoveries by means of molecular genetics allow the description of abnormalities in the development of the pituitary and its ability to produce and secrete GH and other trophic hormones. Classifications systems of GHD may also consider whether GHD is congenital and follows a certain pattern of inheritance or whether it is acquired. 6 ‘Classical’ Idiopathic GHD Comments: Idiopathic GHD (i.e. no aetiology is established) is still the most common form of GHD. It appears trivial, but nevertheless important to note that every reasonable effort should be made to exclude diagnoses other than GHD beforehand. The key diagnostic tools to establish the diagnosis are blood GH measurements collected under spontaneous conditions or after physiological or pharmacological stimuli (e.g. arginine). In order to avoid falsely positive results these tests should be conducted under strict standardisation and under the least stressful conditions for the child. Since it is assumed that test results are prone to be falsely low, at least two independent tests are required to confirm the diagnosis. A cut-off level of 20 mU/l (equivalent to 10, 7.7 or 6.7 μg/l, depending on the standard used; 1 mg = 2, 2.6 or 3 IU) for GH in serum or plasma is generally accepted as a guideline. This cut-off may, however, vary according to the applied GH assay, the test procedures used and the normative references available. It is obvious that all available methods should be applied to increase the likelihood of the diagnosis of GHD. Normal stature, a normal height velocity and a normal bone age are probably not completely excluding the diagnosis of idiopathic GHD, but make its presence quite unlikely. Likewise normal/high-normal IGF-I (IGFBP-3, ALS) levels are also not supportive of the diagnosis. An MRI of the pituitary area is an essential part of the diagnostic process of GHD. A small Short Stature 7 anterior pituitary or a pituitary abnormality are not an a priori proof of a disturbed GH secretion, but are likely to provide evidence that the biochemically established diagnosis of GHD is correct. With greater refinement in our diagnostic arsenal less cases with GHD will be classified idiopathic. 7 Neurosecretory dysfunction (NSD) Comments: The term NSD describes a specific situation, in which GH levels observed during stimulation tests are normal, but spontaneously secreted GH is low. This situation was first described in children who had been treated with radiotherapy to the cranium as part of treatment for a malignancy. In this situation NSD can be seen as a special form of organic GHD. However, this situation was also found to exist in children without such a history. Critics have emphasised that there is a great day-to-day variation in spontaneous GH secretion, and that spontaneous GH secretion is sometimes very low in normally growing children. 8 Bioinactive GH (Kowarski syndrome) Comments: Bioinactive GH can be suspected in children clinically resembling ‘classical’ GH, with low circulating IGF-I levels but with normal GH secretion based on immunoassay measurements. Treatment with GH leads to an increase in IGF-I levels and a growth response to GH. It can be assumed that one reason for bioinactive GH is a congenital structural abnormality of GH, which impairs/abolishes interaction with the GH receptor and subsequent signal transduction. Sophisticated methods (e.g. RRA, bioassay, gene analyses) are required to prove such an entity. Up to 2006, only a few welldocumented cases have been published. 9 Psychosocial deprivation Synonym: Emotional deprivation. Phenotype: Behavioural abnormalities, such as apathy, watchfulness, autoerotic activity, delayed developmental behaviour. Poor growth at home, catch-up growth in hospital or foster home. Reversible GH deficiency has been described. 10 Idiopathic short stature (ISS) Comments: The definition of ISS (previously also called normal variant short stature, or non-GH-deficient short stature) is based on the exclusion of other likely causes of short stature, as well as the following minimal criteria: – normal size at birth (> –2 SDS) for gestational age – normal body proportions – no evidence of chronic organic disease – no psychiatric disorder or severe emotional disturbance – normal food intake – no evidence of endocrine deficiency. The tempo of growth throughout the growth process may either be slow or normal. Reference: Ranke MB: Horm Res 1996;45:64–66. 11 Familial (idiopathic) short stature (FSS) Phenotype: Height for chronological age is below the 2.3rd centile (< –2 SDS) for given age, sex and population, and height is within the ‘normal’ range for parental height. The parent-specific lower limit of height (SDS) can be calculated in various ways. ESPE Classification of Paediatric Endocrine Diagnoses 8 References: Preece MA: Horm Res 1996;45:56–58; Ranke MB: Horm Res 1996;45:64–66; Hermanussen M, Cole J: Horm Res 2003;59:180–183; Wit JM: Horm Res 2007;67(suppl 1):50–57. 12 Familial short stature, onset of puberty not yet known Comments: A short prepubertal child who is not short for target height with an age below the upper limit of the normal age at pubertal onset is classified here. In some cases, a positive family history for pubertal delay and a delayed bone age suggest the syndrome of constitutional delay in growth and adolescence, although in typical cases height in childhood is lower than expected for target height. As soon as the onset of puberty (G2 or B2) is known, the patient can be classified as 1C.1a or 1C.1b. 13 Non-familial (idiopathic) short stature (NFSS) Phenotype: Height for chronological age is below the 2.3rd centile (< –2 SDS) for given age, sex and population and height is below the ‘normal’ range for parental height. Comments: Non-familial short stature (NFSS) is a purely descriptive term. The majority of children with NFSS have a condition usually labelled as constitutional delay in growth and adolescence (CDGA) or small/delay. In CDGA short stature is combined with a retardation of the tempo of growth. When signs of puberty are delayed beyond the normal age range, CDGA can be diagnosed. Before pubertal age, a delay in bone maturity may be suggestive for CDGA, but strictly speaking CDGA can only be diagnosed if puberty is indeed delayed. In these cases the diagnosis NFSS with pubertal onset not yet known is more appropriate. 14 Non-familial idiopathic short stature, onset of puberty not yet known A short prepubertal child who is short for parental height with an age below the upper limit of the normal age at pubertal onset is classified here. In some cases, a positive family history for pubertal delay and a delayed bone age suggest the syndrome of constitutional delay in growth and adolescence. As soon as the onset of puberty (G2 or B2) is known, the patient can be classified as 1C.2a or 1C.2b. Short Stature 9 ESPE Code Diagnosis 2 TALL STATU R E 2A PR I MARY G ROW TH D ISO R D E R S1 2A.1 Clinically defined (dysmorphic) syndromes with sex chromosome abnormality including aneuploidy Syndromes classified elsewhere: 47,XXY (Klinefelter syndrome) [primary 14A.3] 47,XYY syndrome [primary 14A.6] Fragile X syndrome [primary 14B.14; secondary 2A.3a] 47,XXX syndrome2 Other specified X and Y chromosome aneuploidy syndromes3 2A.1a 2A.1b 2A.1y OMIM ICD10 Q97.0 Q97.1 Q97.8 Q98.8 2A.1z Other syndromes, unspecified 2A.2 Dysmorphic syndromes due to metabolic/connective tissue abnormality Syndromes classified elsewhere: Marfan syndrome (14B.20) Marfan-like syndrome, not genetically confirmed Homocystinuria4 Total lipodystrophy (Berardinelli (generalised) lipodystrophy syndrome) [primary 11A.3b.5] Other specified syndromes Other syndromes, unspecified Q97.9 Q98.9 2A.2a 2A.2b 2A.2c 2A.2d 2A.2y 2A.2z 2A.3 2A.3a 2A.3y 2A.3z Other dysmorphic syndromes with symmetrical overgrowth Syndromes classified elsewhere: Bannayan-Riley-Ruvalcaba syndrome (14B.4) Elejalde syndrome (14B.12) Fragile X syndrome (14B.14) Marshall-Smith syndrome (14B.21) Nevo syndrome (14B.23) Simpson-Golabi-Behmel syndrome (14B.32) Sotos syndrome (14B.34) Weaver syndrome (14B.36) Other specified syndromes Other syndromes, unspecified ESPE Classification of Paediatric Endocrine Diagnoses #154700 Q87.4 #154705 E72.1 #236250 E72.1 #236250 E88.1 Q87.3 10 ESPE Code Diagnosis 2A.4z Dysmorphic syndromes with partial/asymmetrical overgrowth Disorders classified elsewhere: Beckwith-Wiedemann syndrome (14B.5) Klippel-Trenaunay-Weber syndrome (14B.17) Proteus syndrome (14B.26) Other specified syndromes Other syndromes, unspecified 2B SECONDARY GROW TH DISORDERS5 2B.1 2B.1c Growth hormone overproduction GH-producing adenoma (solitary) [primary] [secondary 6B.0]6 GH-producing adenoma, as part of McCune-Albright syndrome or MEN1 syndrome GHRH excess7 2B.2 Hyperinsulinism 2B.2a Primary Disorders classified elsewhere: Transient hyperinsulinism (11B.1a) Congenital hyperinsulinism (11B.1b) Other disorders, unspecified Secondary Disorders classified elsewhere: simple obesity (5A) Obesity with acanthosis nigricans 8 hyperlipidaemia and sexual precocity Other disorders, unspecified 2A.4 2A.4a 2A.4y 2B.1a 2B.1b OMIM ICD10 Q87.3 E22.0 #102200 #174800 Q78.1 +131100 E22.0 E15.0, E16.1 2B.2a.1 2B.2a.9 2B.2b 2B.2b.1 2B.2b.2 2B.2b.9 2B.3 Familial (isolated) glucocorticoid deficiency (ACTH insensitivity, hereditary unresponsiveness to ACTH) [primary 8A.2d] 2B.4 Hyperthyroidism8 2B.5 Conditions leading to tall stature in childhood, and normal or short stature in adulthood Tall Stature L83 E16.1 #202200 E05 11 ESPE Code Diagnosis 2B.5a 2B.5y 2B.6y 2C I D I O P AT H I C ( N O R M A L V A R I A N T T A L L S T AT U R E ) 9 2C.1 Genetic (familial, or constitutional) tall stature10 2C.2 Non-familial idiopathic tall stature 2B.6a 2B.6b 2B.6c 1 ICD10 +107910 E34.9 +133430 E34.9 Diagnoses classified elsewhere: Precocious puberty (3A) Exogenous estrogens or androgens (3A.2d) Hyperthyroidism (7B) Congenital adrenal hyperplasia (8A.1) Other specified conditions Conditions leading to normal height in childhood, and tall stature in adulthood Disorder classified elsewhere: Gonadotrophin deficiency (long limbs and hypogonadism) (6A.3) Aromatase deficiency Estrogen receptor dysfunction Other specified conditions 2B.6 OMIM E34.4 Primary growth disorders Causes of tall stature and causes of short stature are subclassified into three groups: primary, secondary and idiopathic. Some clinicians prefer the term ‘dysmorphic syndromes with overgrowth‘ instead of ‘primary growth disorders’. 2 47,XXX syndrome (Triple X syndrome) Phenotype: Tall stature. Increased risk of variable degree of behavioural difficulties and learning disability. Delayed menarche, and premature ovarian failure in some patients. Increased incidence of other congenital anomalies. Ovarian function usually normal but primary ovarian impairment seen in some individuals. Comments: Increased risk of sex chromosome anomaly in child of 47,XXX female, therefore prenatal genetic counselling important. 48,XXXX females are tall with learning disability, radioulnar synostosis, and often incomplete puberty. 3 Other X and Y chromosome aneuploidy syndromes (48,XXXY, 48,XXYY, 49,XXXXY, 49,XXXYY) Phenotype: Phenotype similar to Klinefelter syndrome but with more marked cognitive impairment, more severe hypogonadism, and additional features (e.g. radioulnar synostosis in 48,XXXY and 49,XXXYY). Often associated with hypergonadotrophic hypogonadism. ESPE Classification of Paediatric Endocrine Diagnoses 12 4 Homocystinuria Phenotype: Tall stature with marfanoid habitus and osteoporosis. Learning disability common. Medial degeneration of aorta with frequent arterial and venous thromboses. Myopia and downward subluxation of lens. Homocystine and methionine accumulation interfere in collagen formation resulting in lens and skeletal changes, and platelet thrombosis causing vascular occlusion. 5 Secondary growth disorders Comments: Some clinicians may favour the term ‘tall stature caused by hormone diseases’. 6 Growth hormone overproduction, growth hormone producing adenoma (pituitary gigantism) Phenotype: Inappropriately tall stature for parental heights. Enhanced height velocity. May have headache, visual symptoms (e.g. field defect, optic nerve pallor). Elevated GH levels with failure to suppress below 5 mU/l following oral glucose load. Abnormal overnight GH profile with loss of normal pulsatility, and failure to return to baseline. Enhanced prolactin response to TRH. May show LH, FSH, ACTH and TSH deficiency if there is a mass effect from the adenoma. Comments: Can be associated with McCune-Albright syndrome or MEN type 1. 7 GHRH excess (GHRH-producing tumour) Phenotype: Tall stature/gigantism. Suprasellar/hypothalamic tumour; secondary enlargement of pituitary. No suprasellar/hypothalamic lesion detectable in some cases. GHRH levels raised. 8 Hyperthyroidism Comment: Juvenile hyperthyroidism can lead to tall adult stature, but in most cases adult height is within the normal range. 9 Idiopathic tall stature Comments: The term idiopathic tall stature is used for consistency with the classification of short stature. Some clinicians may favour other terms (e.g. normal variant tall stature), and subclassify children in this category in various ways, e.g. normal genetic tall stature, constitutional advance in growth and adolescence. We prefer the term idiopathic as, even if a condition is familial, its cause is unknown. 10 Genetic tall stature (familial tall stature, constitutional tall stature) Phenotype: Height SDS > +2.0 and height SDS within target range (for definitions, see under familial short stature). Usually there is no significant bone age advancement. Comments: Tall child of tall parents, destined to be a tall adult. Height prediction helpful from 10 years in girls and 11 years in boys. NB: Very tall stature in one parent may reflect a dominant disorder (e.g. Marfan syndrome). Tall Stature 13 ESPE Code Diagnosis OMIM ICD 10 3 D I SO R D E R S O F PU B E R T Y 3A P R E C O C I O U S P U B E R T Y1 E30.1 3A.1 Central precocious puberty2 Due to disorder classified elsewhere: Congenital CNS malformations (6E.1) Acquired CNS disorders (6F), e.g. cerebral tumours, hydrocephalus, post infection, post trauma, post cranial irradiation, neurofibromatosis3 Secondary to peripheral precocity Hypothalamic hamartoma4 Other specified non-CNS disorders Idiopathic5 (including associated with migration from developing countries) E22.8 3A.2 Peripheral precocious puberty6 E30.1 (R) 3A.2a Due to disorder classified elsewhere: Congenital adrenal hyperplasia (8A.1) Virilising and feminising adrenal tumours (8C.2) Leydig cell tumour of the testis (9D.2a) Germ cell tumour of the ovary (10E.4a) Granulosa tumour of the ovary (10E.4b.1) Aromatase deficiency in females (4C.2a) Gonadotrophin secreting tumours7 CNS tumours, e.g. germ cell tumours Other tumours, e.g. choriocarcinoma, hepatoblastoma, or germ cell tumours in the mediastinum Autonomous gonadal function Due to disorder classified elsewhere: McCune-Albright Syndrome (14B.22) Autonomous ovarian follicular cyst Familial testotoxicosis8 Aromatase deficiency in males9 Exogenous sex steroids 3A.1a 3A.1b 3A.1c 3A.1y 3A.1z E22.8 E22.8 E22.8 E22.8 E25 (=) 3A.2b 3A.2b.1 3A.2b.2 3A.2c 3A.2c.0 3A.2c.1 3A.2c.2 3A.2c.3 3A.2d E25.0 E30.1 E30.1 E30.1 #174800 Q78.1 E28.0 E29.0 +107910 drug code Y42 3A.2e 3A.2f Primary hypothyroidism10 Tumours producing androgen or estrogen steroids, not originating from the testis or ovary ESPE Classification of Paediatric Endocrine Diagnoses E03.9 E25.9 14 ESPE Code Diagnosis OMIM ICD 10 3B S I N G L E V A R I AT I O N S O F N O R M A L P U B E R T Y 3B.1 Premature adrenarche11 E27.0 3B.2 E30.8 3B.2b Premature thelarche In infancy (infantile mammoplasia)12 Beyond infancy (thelarche variant)13 3B.3 Premature isolated menarche E30.1 3B.4 Hypertrichosis L68.1– 3B.2a L68.9 Q84.2 3C C O N T R A S E X U A L P U B E R TA L D E V E L O P M E N T 3C.1 Feminisation (males): gynaecomastia Due to disorders classified elsewhere: Primary hypogonadal conditions (hypergonadotrophic hypogonadism) (9A) Partial androgen insensitivity (4B.2d) Feminising adrenal tumours (8C.2) Physiological gynaecomastia Gynaecomastia of the newborn male Adolescent gynaecomastia14 Iatrogenic Neoplasms Increased conversion of androgen to oestrogen imbalance oestrogens/androgens (e.g. XXY) Increased substrate for peripheral aromatase Increase in extraglandular aromatase Idiopathic 3C.1a 3C.1b 3C.1b.1 3C.1b.2 3C.1c 3C.1d 3C.1e 3C.1e.1 3C.1e.2 3C.1e.9 3C.2 3C.2a Virilisation/hirsutism (females)15 Excluded: Virilised infant/ambiguous genitalia (4C) Hypertrichosis (3B.4) Due to disorders classified elsewhere: Congenital adrenal hyperplasia (8A.1) Cushing syndrome (8C.1) Virilising adrenal tumours (8C.2) Polycystic Ovary Syndrome (10B) Germ cell tumour of the ovary (10E.4a) Disorders of Puberty N62 N62 E25.0 N62 E25, L68.0 15 ESPE Code Diagnosis 3C.2z Iatrogenic Other specified disorder Idiopathic 3D D E L AY E D P U B E R T Y 3D.0 Due to disorder classified elsewhere Hypogonadotrophic hypogonadism (6A.4) Hypergonadotrophic hypogonadism: In females (10A) In males (9A) Hyperprolactinaemia (6B.5) Steinert syndrome (myotonic dystrophy) (14B.35) 3C.2c 3C.2y 3D.1a Constitutional delay in (growth and) puberty16 Excluded: Prepubertal (idiopathic) short stature associated with delayed pubertal onset (1C.2b) With positive family history With negative family history Parental puberty onset unknown 3E MENSTRUAL DISORDERS 3E.1 Amenorrhoea Primary amenorrhoea Excluded: Disorders of sex development (4) Due to disorder classified elsewhere: Anatomical defect female genitalia (10F–G) Hypergonadotrophic hypogonadism (10A) Chronic anovulation with oestrogen present Polycystic ovary syndrome (10B) Adrenal disorders, e.g. Cushing (8C.1) Adult-onset congenital adrenal hyperplasia (8A.1c.3) Thyroid disease: hypothyroidism (7A), hyperthyroidism (7B) Ovarian tumour (10E.4) Hypogonadotrophic hypogonadism (6A.4) Hyperprolactinaemia (6B.5) 3D.1 3D.1a 3D.1b 3E.1a 3E.1a.0 ESPE Classification of Paediatric Endocrine Diagnoses OMIM ICD 10 E30.0 E23 E23.0 E30.0 N91.2 N91.0 E28.8 16 ESPE Code Diagnosis 3E.1a.8 3E.1a.9 3E.1b 3E.1b.0 3E.1b.8 3E.1b.9 3E.2 3E.2a 3E.2b Due to other, known causes not classified elsewhere: E.g. growth-retarding diseases such as anorexia nervosa Idiopathic Secondary amenorrhoea Due to disorder classified elsewhere: See 3E.1a.0 Due to other, known causes not classified elsewhere: E.g. growth-retarding diseases such as anorexia nervosa Idiopathic Oligomenorrhoea Primary Secondary OMIM ICD 10 N91.1 N91.5 N91.3 N91.4 3E.3b Abnormal uterine bleeding (excessive, frequent, irregular menses) Dysfunctional uterine bleeding17 Due to structural pelvic pathology, pregnancy or systemic disease 3E.4 Dysmenorrhoea N94.6 3E.5 Premenstrual syndrome N94.3 3E.8 Other, specified menstrual disorder N94.8 3E.3 3E.3a N92 N93.9 1 Precocious puberty Phenotype: Female: Breast development before the age of 8 years. Male: Genital development before the age of 9 years. Both sexes: Acceleration of growth, advancement of bone age. Comments: Data from the United States suggest that sexual precocity should be defined by the onset of secondary sexual development before 6 years in black girls and before 7 years in white girls [Midyett et al., 2003]. This may be associated with the high prevalence of overweight. In Europe the traditional age limits have been maintained by most clinicians. Reference: Midyett LK, et al: J Pediatr 2003;111:47–51. 2 Central precocious puberty (CPP) Synonyms: True precocious puberty, gonadotrophin-dependent precocious puberty. Phenotype: Pubertal levels of LH, FSH, and E2 or T; pubertal response to GnRH test. Comments: Pubertal development in CPP is often not only early, but also more rapidly progressive than observed in normally timed pubertal development. In most girls (90%) with CPP intensive diagnostic investigation will reveal no underlying causes and therefore is called ‘idiopathic’. GnRH analogs are effectively suppressing gonadotropin secretion, but do not postpone central development of the GnRH pulse generator. In most boys with CPP a CNS disorder can be detected. Disorders of Puberty 17 3 Acquired CNS disorders Comments: Various intracranial processes, including increased intraventricular pressure, can induce maturation of the GnRH pulse generator. The mechanisms are unknown. TGF-α may play a role (hamartomas). Infections of the central system at a young age may be associated with CPP in later life. Cranial irradiation may induce hypothalamic-pituitary deficiencies, but also CPP and early puberty. 4 Hamartoma Comments: Presumably as a result of pressure on the hypothalamic area, a cerebral tumour may initiate maturation of the GnRH pulse generator. Glial-derived transforming growth factor (TGF-α) may contribute to the neuro-endocrine mechanisms. Hamartoma may contain GnRH neurons functioning as an ectopic source of GnRH. 5 Idiopathic central precocious puberty Phenotype: In girls with start of breast development just before the CPP age limit, one should consider borderline early puberty. In girls with start of breast development, isolated or associated with some other pubertal signs without evidence of CPP, thelarche variant is considered (see 3B.2b). In boys, idiopathic CPP is rare (10%). Comments: Children adopted from developing countries and moved to a more affluent environment have an increased incidence of early and precocious puberty. Reference: Parent AS, et al: Endocr Rev 2003;24:668–693. 6 Peripheral precocious puberty (PPP) Synonyms: Pseudoprecocious puberty, gonadotrophin-independent precocious puberty Phenotype: Female: breast development before the age of 8 years. Male: Genital development and/or virilisation before the age of 9 years. Both sexes: Acceleration of growth, advancement of bone age. Pubertal levels of sex steroids, low gonadotrophin levels. Comments: In PPP, development of the sex characteristics is the result of gonadotrophin independent steroid production. In boys, the major cause is late onset 21 hydroxylase deficiency; in case of a familial form PPP may be the result of an activating mutation of the LH receptor (3A.2c.2). In girls, PPP is often the result of autonomous ovarian cysts; the underlying cause may be ovary stimulating antibodies and activating mutations of the FSH receptor. 7 HCG-producing tumours Comments: These tumours are classified by some under the heading of gonadotrophin-dependent precocious puberty [e.g. Brook CGD, et al (eds): Clinical Pediatric Endocrinology, ed 5, 2005]. If gonadotrophin-dependent precocious puberty is defined as being caused by premature maturation of an otherwise normal hypothalamic-pituitary system, HCG-producing tumours have a more logical place in the group of gonadotrophin-independent precocious puberty. 8 Familial testotoxicosis Synonym: Leydic cell hyperplasia. Phenotype: Development of sex characteristics as a result of premature maturation of the testis but hardly any testis growth. FSH (–), LH (–), T (+) caused by constitutive activation of LH receptor. Comment: Pubertal signs such as virilisation and an increased height velocity start at the age of 1–4 years. ESPE Classification of Paediatric Endocrine Diagnoses 18 09 Aromatase deficiency Phenotype: Tall stature due to failure of epiphyseal closure at puberty. Elevated androgen levels causing virilisation but not skeletal maturation in girls. Cliteromegaly, with polycystic ovaries in pubertal females. Normal puberty in males with tall stature, eunuchoid habitus and osteoporosis in adulthood. T (+), DHT (+), adione (+), LH (+), FSH (+), E1 (–), E2 (–). 10 Primary hypothyroidism Comments: In some patients with primary hypothyroidism FSH are increased. Furthermore, TSH has weak agonist properties at the FSH receptor. 11 Premature adrenarche Synonym: The term premature pubarche is often used for this condition. However, strictly speaking premature pubarche means early onset of pubic hair, independent of the cause. Premature adrenarche is the most frequent subgroup, diagnosed if other causes of premature pubarche (e.g. late-onset congenital adrenal hyperplasia) are excluded. Phenotype: Premature pubarche (pubic hair before the age of 8 years) and premature axillary hair, and sweating before the age of 8 years; none or slight acceleration of growth and of bone age. No other sex characteristics. There may be an increased prevalence of functional ovarian hyperandrogenism in mid-teenage years. Pubertal or adult levels of DHEA (–S). 12 Premature thelarche in infancy Phenotype: Premature breast development without other signs of puberty; no or slight acceleration of growth and of bone age; mostly in infancy (60% between 6 months and 2 years). Onset is rare after 4 years. Most regress 6 months to 6 years after diagnosis. Incomplete suppression of GnRH axis, FSH (+), LH (N), E2 (N). Comment: The GnRH test in a girl with premature thelarche will elicit a high response of FSH and low response of LH in contrast to girls with CPP with exaggerated responses of especially LH and FSH. 13 Premature thelarche beyond infancy Synonym: Thelarche variant. Phenotype: A slowly progressive variant of precocious puberty in girls, characterised by the start of breast development between the age of 7 and 8 years. The progression is slow. Breast development is often associated with mild or partial signs such as slight growth acceleration and/or mild advancement of bone age. This state will gradually transform into a normal pubertal development at an appropiate age. 14 Adolescent gynaecomastia Phenotype: Breast development in pubertal boys. E2, T, LH and FSH in early to mid-pubertal range. By increasing T levels, the gynaecomastia usually disappears. Comment: Galactorrhoea is extremely rare in adolescents with gynaecomastia. 15 Hirsutism Phenotype: Excessive growth of terminal hair on the face and body of a woman in a typical male pattern distribution. Assessment of the amount, distribution, and severity of hirsutism is by the method of Ferriman-Gallwey. Disorders of Puberty 19 Comment: Causes include: (1) excess androgen production by the ovary and/or the adrenal; (2) rise in the level of biologically active free androgen, and/or (3) increased sensitivity of the hair follicle to androgens. Reference: Ferriman D, Gallwey JD: J Clin Endocrinol Metab 1961;21:1440–1447. 16 Constitutional delay in (growth and) puberty Synonyms: Constitutional delayed puberty, familial delay of growth and adolescence, constitutional delay of growth and adolescence. Phenotype: First signs of puberty after the age of 13 in girls and 14 in boys. Comment: More common in boys than in girls. 17 Dysfunctional uterine bleeding Comments: Although usually anovulatory, may also be ovulatory. To be classified as dysfunctional uterine bleeding it must not be associated with structural pelvic pathology, pregnancy or systemic disease. ESPE Classification of Paediatric Endocrine Diagnoses 20 ESPE Code Diagnosis 4 4A OMIM ICD 10 D I SO R D E R S O F S E X DEVE LO PM E NT (DSD) 1 SEX CHROMOSOME DISORDERS OF SEX DEVELOPMENT Excluded: Disorders of gonadal differentiation that do not result in sex reversal/virilised female infant/undervirilised male such as: Klinefelter syndrome (14A.3) Turner syndrome (14A.5) 46 XX gonadal agenesis/dysgenesis (10A.1b/c). For later onset virilisation see contrasexual pubertal development (3C) 4A.0 Disorders of gonadal differentiation classified elsewhere Code 4A.0 is only to be used with a corresponding primary code (in parentheses) if the individual is undervirilised and/or has ambiguous genitalia; if genitalia are normal then use primary code alone or together with code for hypergonadotrophic hypogonadism (9A.0) as indicated Disorders classified elsewhere: 45,X (Turner syndrome and variants) (14A.5) 47,XXY (Klinefelter syndrome and variants) (14A.3) 46 XX gonadal agenesis/dysgenesis (10A.1b/c) 4A.1 45,X/46,XY (mixed gonadal dysgenesis, ovotesticular DSD)2 Only includes individuals with 45X/46XY (or similar forms of sex chromosome mosaicism, e.g. 45,X/46,XY/47,XYY) and asymmetric gonads such as dysgenetic testes on one side and streak on other, or ovotestis on one side with streak on other; individuals with bilateral dysgenetic testes or dysgenetic testes in association with normal male or female genitalia are not classified here but classified under 4A.0 Q97.8 4A.2 46,XX/46,XY (chimeric, ovotesticular DSD)3 Q99.0 4A.8 Other, specified forms of sex chromosome DSD causing abnormal genitalia Q99.8 Disorders of Sex Development (DSD) (female phenotype) or Q98.8 (male phenotype) 21 ESPE Code Diagnosis 4B OMIM ICD 10 +306100 Q97.3 4 6 , X Y D I S O R D E R S O F S E X D E V E L O P M E N T4 Excluded: Hypospadias (9E.1) or cryptorchidism (9B) without clear underlying endocrine etiology, and Leydig cell hypoplasia if not leading to a DSD (9A.1b) 4B.0 Undervirilised male classified elsewhere Note: this is a supplementary code to be used together with the primary code from respective chapters 4B.1 Disorders of gonadal (testicular) development Complete gonadal dysgenesis (Swyer syndrome)5 WT1; WAGR, Frasier syndrome and Denys-Drash syndrome 4B.1a 4B.1a.1 *607102 #136680 #194080 4B.1a.2 4B.1a.3 4B.1a.4 4B.1a.5 4B.1a.6 4B.1a.7 4B.1a.8 4B.1a.9 4B.1a.10 4B.1a.88 4B.1a.99 4B.1b 4B.1c 4B.1d 4B.2 4B.2a 4B.2b 4B.2b.1 4B.2b.2 SF1 SRY SOX-9 (campomelic dysplasia) DHH ATRX ARX DMRT1 DAX1 WNT4 Other specified causes Other causes, unspecified Partial gonadal dysgenesis Gonadal regression Ovotesticular DSD (includes 46,XY true hermaphroditism) Disorders in androgen synthesis or action Disorders classified elsewhere: Enzyme defects affecting synthesis of both corticosteroids and testosterone are primarily classified under ‘congenital adrenal hyperplasia’ (8A.1): Lipoid CAH (cholesterol side chain cleavage deficiency, P450scc) (8A.1a) 3beta-Hydroxysteroid dehydrogenase deficiency (8A.1b) 17alpha-Hydroxylase and 17/20 lyase deficiency (P450c17) (8A.1f) P450 oxidoreductase deficiency (8A.1g) Smith-Lemli-Opitz syndrome (14B.33) Androgen biosynthesis defect 17-Hydroxysteroid dehydrogenase III deficiency6 5␣-Reductase deficiency7 ESPE Classification of Paediatric Endocrine Diagnoses +184757 *480000 *608160 *605423 *300032 *300382 *602424 #300018 *603490 Q99.1 Q99.1 #235600 Q99.1 E29.1 #264300 E34.5 *184753 E34.5 22 ESPE Code Diagnosis 4B.2c 4B.2d 4B.2d.1 4B.2d.1a 4B.2d.1b 4B.2d.2 4B.2d.2a 4B.2d.2b 4B.3 4B.3a 4B.3b OMIM ICD 10 LH receptor defects, e.g. Leydig cell hypoplasia, aplasia8 +152790 Defect in androgen action (androgen insensitivity syndromes)9 #300068 Complete androgen insensitivity syndrome (CAIS) – CAIS with demonstrated mutation of the androgen receptor gene – CAIS without demonstrated mutation of the androgen receptor gene Partial androgen insensitivity syndrome (PAIS) – PAIS with demonstrated mutation of the androgen receptor gene – PAIS without demonstrated mutation of the androgen receptor gene E29.1 Disorders of AMH and AMH receptor (persistent Müllerian duct syndrome) – Synonym: Hernia uteri inguinale, female genital ducts in otherwise normal male Defect in the gene for MIS10 Defect in the gene for MIS (type II) receptor11 #261550 Q56.1 *600957 Q56.1 *600956 Q56.1 E34.5 4B.4 Iatrogenic impairment of masculinisation of foetus (specify agent) Q56.1 4B.8 Due to other, known causes not classified elsewhere E.g. severe hypospadias, cloacal extrophy E29.8 4B.9 Idiopathic E29.9 4C 4 6 , X X D I S O R D E R S O F S E X D E V E L O P M E N T 12 Excluded: Gonadal dysgenesis (10E.1) 4C.0 Virilised female classified elsewhere Congenital adrenal hyperplasia (8A.1) 4C.1 Disorders of gonadal (ovarian) development Ovotesticular DSD (previously called 46,XX true hermaphroiditism) Testicular DSD13 SRY+14 dup SOX915 4C.1a 4C.1b 4C.1b.1 4C.1b.2 Disorders of Sex Development (DSD) Q99.1 278850 Q99.1 *480000 *608160 23 ESPE Code Diagnosis OMIM ICD 10 #201750 E25.8 4C.2c.5 Androgen excess Virilisation due to androgen production by the foetus and/or placenta Disorders classified elsewhere: 3beta-Hydroxysteroid dehydrogenase deficiency (8A.1b) 21-Hydroxylase deficiency (P450c21) (8A.1c) 11beta-Hydroxylase deficiency (8A.1d) Glucocorticoid receptor defect (8A.1h) Foetoplacental (P450-aromatase deficiency16, P450 oxidoreductase deficiency) Maternal Iatrogenic (specify agent) Virilising ovarian tumour Virilising luteoma of pregnancy Virilising adrenal tumour Congenital virilising adrenal hyperplasia in mother 4C.8 Due to other, known causes not classified elsewhere E25.0 4C.9 Idiopathic virilisation of the foetus E25.0 4D UNCLASSIFIED FORMS OF ABNORMAL SEXUAL D E V E L O P M E N T/ A N AT O M I C A L D I S R U P T I O N S 4D.1 Ambiguous genitalia with multiple congenital anomalies In the 46,XY individual In the 46,XX individual 4C.2 4C.2a 4C.2b 4C.2c 4C.2c.1 4C.2c.2 4C.2c.3 4C.2c.4 4D.1a 4D.1b E25.0 D39.1 D27 D35.0 E25.0 Q56.4 1 General commentary The LWPES/ESPE Consensus Group proposed the term ‘Disorders of sex development’ (DSD), as defined by congenital conditions in which development of chromosomal, gonadal, or anatomical sex is atypical. Synonyms: Previously used terms: intersex disorders, genital abnormalities [Hughes IA, et al: Arch Dis Child 2006;91:554]. 2 Mixed gonadal dysgenesis (MGD) Phenotype: Variable degree of impaired masculinisation, from almost female (in case of complete female phenotype classify as Turner syndrome (14A.5)) to normal male genitalia (14A.4). Hypospadias, cryptorchidism may be present. At puberty FSH and LH are usually elevated and testosterone decreased. Secondary to sex chromosomal mosaicism in a male, the gonads have become dysgenetic in combinations ovary/testis, or testis/streak. Sometimes a tumour is found instead of a gonad. ESPE Classification of Paediatric Endocrine Diagnoses 24 Comments: The term MGD can be used when the above chromosomal mosaicism (or, in rare cases, 45,X/46,XY/47,XYY) is found, even if gonadal dysgenesis has not been proven anatomically. 3 Ovotesticular DSD Synonym: This condition was previously called ‘true hermaphrodite’. Phenotype: Baby born with ambiguous genitalia. Hypospadias, undescended testes/clitoromegaly, labial fusion. Both testicular and ovarian tissues identified in the gonads. T variable, FSH (+/N), LH (+/N). 4 46,XY DSD Synonyms: Was previously called ‘male pseudohermaphrodite’, or ‘undervirilisation of an XY male’, or ‘undermasculinisation of an XY male’. Phenotype: Incomplete maculinisation, hypospadias, cryptorchidism, bifid scrotum. Comments: The diagnosis ‘incomplete masculinisation’ is really a symptom that may be caused by several different diseases. There is no sharp line between 46,XY DSD and ‘hypospadias’. Generally, the former term is used when at any time there has been uncertainty about the sex of rearing. 5 46,XY complete gonadal dysgenesis Synonym: Previously called ‘XY sex reversal’. Phenotype: Absence of female puberty. Streak gonads, prepubertal female phenotype. No breast development, primary amenorrhoea. From puberty FSH (+), LH (+), E2 (–). 6 17-Hydroxysteroid dehydrogenase III deficiency Synonym: 17-ketosteroid reductase deficiency (17-KSR deficiency). Phenotype: Female or ambiguous external genitalia, hypospadias, defective virilisation in XY individual. Partial androgenisation at puberty. Defective intrauterine virilisation of male genitalia. Adione (+), T (–), DHT (–), E1 (+), E2 (–), possibly revealed after hCG stimulation. Adione to T ratio (+). 7 5alpha-Reductase deficiency Phenotype: Ambiguous genitalia or micropenis in XY individuals. Partial androgenisation at puberty. Defective intrauterine virilisation of male external genitalia. T (+), DHT (–), possibly revealed after hCG stimulation. T to DHT ratio (+). 8 Leydig cell hypoplasia Synonym: LH receptor deficiency. Phenotype: Ambiguous genitalia. Defective development of Wolffian structures and male external genitalia. LH (+), T (–). 9 Defect in androgen action (androgen insensitivity syndromes) Synonyms: Reifenstein syndrome; complete or partial AIS (CAIS or PAIS). Phenotype: External genitalia varying between female, ambiguous, micropenis or normal male. Hypospadia, cryptorchidism, defective virilisation at puberty; testes small to normal sized in adulthood. Histology shows quantitative and qualitative defect of spermatogenesis. Adulthood: FSH (N/+), LH (N/+), T (N/+). Disorders of Sex Development (DSD) 25 10 Defect in the gene for MIS Phenotype: Persistent Müllerian duct syndrome. Normal male phenotype. 11 Defect in the gene for MIS (type II) receptor Phenotype: Persistent Müllerian duct syndrome. 12 46,XX DSD Synonym: Was previously called female pseudohermaphrodite, or overvirilisation of an XX female, or masculinisation of an XX female. 13 46,XX testicular DSD Synonym: Was previously called ‘XX male’ or ‘XX sex reversal’. 14 SRY+ Phenotype: External genitalia male or ambiguous. Testis or ovotestis. No Müllerian structures. Comment: Translocation of SRY. 15 dup SOX9 Phenotype: External genitalia male or ambiguous. 16 P450-aromatase deficiency Phenotype: In XX individuals ambiguous genitalia. Maternal androgenisation during pregnancy, absent breast development at puberty, except in partial cases. ESPE Classification of Paediatric Endocrine Diagnoses 26 ESPE Code Diagnosis 5 OVE RWE I G HT AN D O B E S IT Y 5A ‘ S I M P L E ’ O B E S I T Y, C O M M O N O B E S I T Y 1 5A.1 5A.1b Apparently prevailing environmental factors Diet/activity balance Emotional environment 5A.2 Apparently prevailing genetic factors 5B GENETIC CAUSES 5B.1 Monogenic obesity syndromes Congenital leptin deficiency2 Leptin receptor deficiency3 Pro-opiomelanocortin (POMC) deficiency4 Prohormone convertase 1 deficiency (if gonadotrophin deficiency is present, also code as 6A.3b.4) MC4R deficiency5 Other specified monogenetic syndromes 5A.1a 5B.1a 5B.1b 5B.1c 5B.1d 5B.1e 5B.1y 5B.2 5B.2a 5B.2y 5B.2z 5B.3 5B.3a 5B.3y 5B.3z Genetic defects associated with obesity Syndromes classified elsewhere: Albright’s hereditary osteodystrophy (pseudohypoparathyroidism) (14B.2) Alström syndrome (14B.3) Carpenter syndrome (14B.7) Cohen syndrome (14B.8) Laurence-Moon-Biedl syndrome (14B.18) Prader-Willi-Labhart syndrome (14B.25) Other specified syndromes Other syndromes, unspecified Chromosomal disorders associated with obesity Syndromes classified elsewhere: Down syndrome (14A.2) Klinefelter syndrome (14A.3) Turner syndrome (14A.5) Other specified syndromes Other syndromes, unspecified Overweight and Obesity OMIM ICD10 E66 E66.0 *601665 E66.8 E66.8 *164160 *601007 #609734 #600955 *155541 *601665 E66.8 E66.9 E66.8 E66.9 27 ESPE Code Diagnosis 5C ENDOCRINE DISORDERS 5C.0 Disorders classified elsewhere Hypothyroidism (7A) Growth hormone deficiency (1B.3) Cushing syndrome (8C.1) Insulinoma (11B.1b.7) Diabetes mellitus inappropriately managed (e.g. insulin omission) (11A), including Mauriac syndrome (14C.2) Polycystic ovary syndrome (10A.2a) 5C.8 Other specified disorders 5D CENTR AL NERVOUS SYSTEM CAUSES (CENTR AL OBESIT Y) 5D.0 Disorders classified elsewhere Cranio-cerebral trauma (6F.4b) Post-neurosurgical lesions (6F.4a) Intercranial space occupying lesions (6F.1) Meningitis/encephalitis (6F.3) Infiltration (6F.2) OMIM ICD10 E66.8 T90 G97.9 D33 G09 G99 5D.8 Other specified disorder 5E O B E S I T Y A C C O M P A N Y I N G I M M O B I L I T Y, M E N TA L D I S T U R B A N C E S , S O C I A L A N D C U LT U R A L P R E S S U R E 5E.1 Muscular dystrophy6 #310200 G71.0 5E.2 Spina bifida with myelomeningocele7 and other #182940 Q05 5E.3 Immobilisation after trauma and/or orthopaedic surgery, etc. 8 T88 5E.4 Mental retardation9 F70–79 5E.5 Psychological/psychiatric disturbances (bulimia)10 F50.2 5E.8 Other specified disorders E66.8 5E.9 Other disorders, unspecified E66.9 ESPE Classification of Paediatric Endocrine Diagnoses E66.8 28 ESPE Code Diagnosis 5F I AT R O G E N I C O B E S I T Y D U E T O M E D I C AT I O N 5F.1 Corticosteroids11 5F.2 Sodium valproate12 5F.3 Insulin13 5F.4 Phenothiazine tricyclate14 5F.5 Cyproheptadine15 5F.8 Other medication, specified OMIM ICD10 E66.1 1 Obesity Synonyms: Severe overweight, fatness. Phenotype: Obesity is an excessive storage of body fat, relative to lean body mass. As a criterion various parameters such as a body mass index or weight-for-height and different cut-off limits (e.g. >P95) are used. A disadvantage of using percentiles of current growth studies is that cut-off levels are changing along with the world-wide trend toward obesity. Therefore, a time- and place-independent cut-off limit has been proposed for obesity, defined as the SDS level that ends at a BMI of 30 at 18 years of age. A similar line is drawn for overweight, ending at a BMI of 25 at 18 years of age [Cole TJ, et al: BMJ 2000;320:1240]. Obese children are frequently taller than their age- and sex-matched peers. Bone age is usually advanced and puberty may occur earlier. Alterations in endocrine functions are thought to be consequences of the obese state: leptin (+), insulin (+), insulin resistance, glucagon (–), growth hormone (–), cortisol (N), cortisol secretion rate (N/–), DHEAS (+), epinephrine and norepinephrine (N/+), IGF-I (N/+), PRL (basal (+), after stimulation (–)). 2 Congenital leptin deficiency Phenotype: Severe early-onset obesity, hyperphagia, metabolic, neuroendocrine and immune dysfunction. Beneficial effects of treatment with recombinant leptin. Increased mortality especially in early childhood. Comment: Mutations in leptin gene. 3 Leptin receptor deficiency Phenotype: Morbid obesity, hyperphagia, hypogonadotrophic hypogonadism, disturbed immune function. TSH (–), GH (–). Clinical features less severe than in patients with congenital leptin deficiency. Comments: Mutations in leptin receptor gene. Prevalence 3% in early onset severe obesity. 4 Pro-opiomelanocortin (POMC) deficiency Phenotype: Congenital deficiency of POMC results in a syndrome of severe obesity, hypoadrenalism and altered skin and hair pigmentation. Leptin (+), cortisol (–). Overweight and Obesity 29 Comments: Mutations of POMC gene on chromosome 2p22. Heterozygote condition and POMC polymorphisms show association with obesity. 5 Melanocortin 4 receptor (MC4R) mutations Phenotype: Most relevant monogenic cause for extreme obesity. Frequently binge eating. Comments: Mutations in MC4R gene. The severity of functional alterations of the mutated MC4R correlates with the onset and severity of obesity. 6 Muscular dystrophy Phenenotype: Duchenne muscular dystrophy is often associated with obesity due to reduction in physical activity. Therapeutic trials with corticoids may aggravate the situation. 7 Myelomeningocele Phenotype: Risk of developing obesity by hypoactivity, subnormal fitness and reduced dynamic activities, especially in non-ambulant individuals. 8 Immobilisation after trauma or surgery Phenotype: Lack of physical activity and energy expenditure may promote weight gain. Indication for early physiotherapeutic intervention. 9 Mental retardation Phenotype: Obesity may be due to hypothalamic dysfunction or lack of understanding adequate food ingestion. Mental retardation may aggravate the situation in syndromes where obesity is already a component of the disease (e.g. Prader-Willi syndrome). 10 Psychiatric disturbances, bulimia Phenotype: Psychiatric disorder with loss of control over eating while consuming large amounts of food. Mostly in young women. Multidisciplinary therapeutic approach including psychotherapy. 11 Corticosteroids Phenotype: In fat cells, corticosteroids increase lipolytic enzymes, resulting in hyperlipidaemia, hypercholesterolaemia and redistribution of fat with truncal obesity and moon facies. 12 Sodium valproate Phenotype: Valproate treatment is associated with obesity, increased fasting insulin levels, hypertriglyceridaemia and hyperandrogenism. May interfere with insulin metabolism in the liver. 13 Insulin Phenotype: Chronic hyperinsulinaemia is associated with obesity by increased ingestion of carbohydrates to prevent hypoglycaemia. 14 Phenothiazine tricyclate Phenotype: Used in psychiatric diseases. Broad spectrum of side effects including weight gain. 15 Cyproheptadine Phenotype: Increases appetite through its antiserotoninergic effect on 5-HT2 receptors in the brain. Leptin (+). ESPE Classification of Paediatric Endocrine Diagnoses 30 ESPE Code Diagnosis 6 PITU ITARY, HYPOTHAL AMUS , CE NTR AL N E RVOUS SYSTE M (CNS) SECTION 1 FUNCTIONAL CLASSIFICATION 1 6A D E F I C I E N C I E S O F A N T E R I O R P I T U I TA R Y HORMONES 6A.0 Disorders classified elsewhere Growth hormone deficiency (1b.3) 6A.1 ACTH deficiency Congenital isolated ACTH deficiency2 Congenital ACTH deficiency in combination with other pituitary deficiencies3 Acquired hypothalamic-pituitary ACTH deficiency, e.g. by longterm glucocorticoid therapy4 6A.1a 6A.1b 6A.1c 6A.2 6A.2a 6A.2a.1 TSH deficiency Congenital isolated TSH deficiency5 – Known genetic defect (TSH-beta, TRHR, TRH, other)6 OMIM ICD10 E23.0 #201400 E03.9 E23.0 #275100 E23.0 +188545 +275120 6A.2a.2 6A.2b 6A.2b.1 6A.2b.2 6A.2c 6A.3 6A.3a 6A.3a.1 – Unknown origin Congenital TSH deficiency in combination with other pituitary deficiencies7 – Secondary (pituitary) hypothyroidism8 – Tertiary (hypothalamic) hypothyroidism9 Acquired hypothalamic-pituitary hypothyroidism10 E23.0 Gonadotrophin deficiency (hypogonadotrophic hypogonadism) X-linked inheritance – Isolated hypogonadotrophic hypogonadism and anosmia (X-linked form of Kallmann syndrome) (KAL1 mutation); [primary] [secondary 14B.16]11 E23.0 Pituitary, Hypothalamus, Central Nervous System (CNS) E23.0 E23.0 E23.0 E23.0 E23.0 +308700 E23.0 31 ESPE Code Diagnosis 6A.3a.2 – Isolated hypogonadotrophic hypogonadism and adrenal hypoplasia congenita (DAX1 mutation) (also classified as 8A.2a.1)12 6A.3b Autosomal inheritance: autosomal forms of Kallmann syndrome – Inactivating mutation of FGFR1 (KAL2)13 – Inactivating mutation of PROKR2 (KAL3)14 – Inactivating mutation of PROK2 (KAL4) – Inactivating mutation of GnRHR15 – GPR54 mutation16 – KISS mutation17 – Prohormone convertase 1 mutation18 – Leptin 1 or leptin receptor mutation19 6A.3b.1 6A.3b.2 6A.3b.3 6A.3b.4 6A.3b.5 6A.3b.6 6A.3b.7 6A.3b.8 OMIM ICD10 E23.0 E23.0 #147950 244200 610628 *138850 E23.0 *604161 E23.0 *603286 E23.0 600955 E23.0 *164160 E23.0 *601007 6A.3b.9 6A.3b.10 6A.3b.11 6A.3b.88 6A.3c 6A.3c.1 – LHR mutation (fertile eunuch syndrome) – Isolated LH deficiency20 – Isolated FSH deficiency21 – Other specified gene mutations, including NELF, etc. Hypogonadotrophic hypogonadism in combination with other pituitary deficiencies22 – Known genetic defect, e.g. PROP1, LHX3, HESX123 #228300 +152780 E23.0 #229070 E23.0 *608137 E23.0 E23.0 *601538 E23.0 *600577 *601802 6A.3c.2 6A.3d 6A.3z 6A.4 6A.4a 6A.4b 6A.4b.1 6A.4b.2 6A.4b.8 6A.4b.9 – Unknown genetic defect Hypogonadotrophic hypogonadism in combination with dysmorphic syndromes, e.g. Prader-Willi-Labhart syndrome [primary 14B.25], Rieger syndrome [primary 14B.28] Hypogonadotrophic hypogonadism, isolated, unspecified Prolactin deficiency Isolated prolactin deficiency24 Prolactin deficiency in combination with other pituitary deficiencies POU1F1 mutation25 PROP1 mutation26 Other specified gene mutations Unknown origin ESPE Classification of Paediatric Endocrine Diagnoses E23.0 E23.0 E23.0 264110 E23.0 E23.0 +173110 E23.0 *601538 E23.0 E23.0 E23.0 32 ESPE Code Diagnosis 6B O V E R P R O D U C T I O N O F A N T E R I O R P I T U I TA R Y HORMONES 6B.0 Disorders classified elsewhere ACTH-producing adenoma (Cushing’s disease) (8C.1) Growth hormone-producing adenoma (synonyms: acromegaly, pituitary gigantism) (2B.1) 6B.1 TSH-producing adenoma27 6B.2 Gonadotrophin-producing adenoma28 6B.3 Prolactin overproduction Prolactinoma29 Hyperprolactinaemia of other cause (e.g. pituitary stalk lesion, primary hypothyroidism)30 6A.3a 6A.3b 6C CENTRAL DIABETES INSIPIDUS [primary 13A.1] 6D HY P OTHAL A M I C DYS FU N C TI O N , N OT CLASSIFIED ELSEWHERE 6D.0 Disorders classified elsewhere Obesity (5D) 6D.8 Other specified functional changes 6D.9 Other functional changes, unspecified SECTION 2 AETIOLOGICAL CLASSIFICATION 1 6E C O N G E N I TA L D I S O R D E R S 6E.1 Congenital CNS malformations Septo-optic dysplasia [primary]31 [secondary 14B.30] Other midline defects, e.g. cleft palate, central maxillary incisor syndrome, EEC syndrome (ectodactyly-ectodermal dysplasiaclefting syndrome)32 6E.1a 6E.1b Pituitary, Hypothalamus, Central Nervous System (CNS) OMIM ICD10 E05.9 M8271/0 E22.1 E23.3 Q04 #182230 Q04.4 Q04.8 33 ESPE Code Diagnosis 6E.1c 6E.1d 6E.1z Ectopic neurohypophysis, absent infundibulum and hypoplastic adenohypophysis33 Hamartoma34 Other congenital CNS malformations, unspecified 6E.2z Congenital hypothalamic-pituitary disorders associated with syndromes Disorders classified elsewhere: Prader-Willi(-Labhart) syndrome (14B.25) Rieger syndrome (14B.28) Chromosomal disorders35 Chromosomal instability syndromes36 Empty sella syndrome37 Other specified syndromes Other syndromes, unspecified 6F ACQUIRED DISORDERS 6F.1 Neoplasms Tumours of the pituitary/hypothalamic region Craniopharyngioma38 Nonfunctional pituitary adenomas39 Other benign structures40 Isolated glioma41 Glioma as part of neurofibromatosis I (von Recklinghausen’s disease, 14B.27) Germinoma, dysgerminoma42 Leukaemia, lymphoma43 Other neoplasms, specified Tumours outside the pituitary/hypothalamic region Pinealoma44 Isolated glioma45 Glioma as part of neurofibromatosis I (von Recklinghausen’s disease, 14B.27) Germinoma, dysgerminoma Medulloblastoma46 Leukaemia, lymphoma Other specified neoplasms 6E.2 6E.2a 6E.2b 6E.2c 6E.2d 6E.2y 6F.1a 6F.1a.1 6F.1a.2 6F.1a.3 6F.1a.4 6F.1a.5 6F.1a.6 6F.1a.7 6F.1a.8 6F.1b 6F.1b.1 6F.1b.2 6F.1b.3 6F.1b.4 6F.1b.5 6F.1b.6 6F.1b.8 6F.2 6F.2a Inflammatory/infiltrative Langerhans cell histiocytosis47 OMIM ICD10 Q04.8 241800 Q85.9 Q04.9 C71.9 604856 D76.0 D76.3 ESPE Classification of Paediatric Endocrine Diagnoses 34 ESPE Code Diagnosis 6F.2b Systemic lupus erythematodes48 6F.2c Neurosarcoidosis49 Lymphocytic neurohypophysitis50 Haemochromatosis51 OMIM ICD10 #601744 #152700 6F.2d 6F.2e 6F.3 6F.3a 6F.3b 6F.3c 6F.3d 6F.4 6F.4a 6F.4b 6F.4c 6F.5 6F.5a 6F.5b Infectious Meningitis52 Encephalitis Abscess of pituitary53 Congenital infection54 Traumatic injury CNS surgery55 Head trauma56 Hypoxic injury57 Iatrogenic Irradiation58 Drugs, e.g. chemotherapy59 6F.6c Secondary to psychiatric disorders Anorexia nervosa60 Emotional deprivation61 Other (specified) 6F.7 Idiopathic 6F.6 6F.6a 6F.6b #181000 #602390 G00 G04, G05 G06.0 G09 G97 S06 G97.8 T66 E23.1 F50.0 1 Classification Comments: Diagnoses are classified according to function and aetiology. In many cases, therefore, two codes will be used for a patient with a CNS disorder. 2 Congenital isolated ACTH deficiency Phenotype: Rare inherited disorder which may cause hypoglycaemia, Addisonian crisis and death in neonates. In pregnant women isolated low oestriol levels may indicate the disease in the foetus. ACTH (–), cortisol (–). Comment: Autosomal-recessive inherited mutations in the TBX19 gene encoding a transcription factor important for differentiation of pituitary corticotroph cells is found in approximately 50%. 3 Congenital ACTH deficiency in combination with other pituitary deficiencies Phenotype: Congenital panhypopituitarism, mostly caused by hypoplasia or aplasia of the pituitary gland due to vascular damage. Neonatal symptoms of Addisonian crisis (hypoglycaemia, hypona- Pituitary, Hypothalamus, Central Nervous System (CNS) 35 traemia), hypothyroidism, growth retardation may occur in association with other brain malformations. ACTH (–), cortisol (–), TSH (–), T4 (–), GH (–). Comments: Mutations in transcription factors responsible for pituitary development. In PROP1 mutations evolving ACTH (and cortisol) deficiency due to progressive pituitary degeneration. 4 Acquired hypothalamic-pituitary ACTH deficiency Phenotype: Fatigue, weakness, hypopigmentation, finally Addisonian crisis. Comments: Can occur in chronic infectious diseases (HIV). Side effect of oral, topical or inhaled longterm glucocorticoid therapy (negative feedback on ACTH). ACTH (–), cortisol (–). 5 Isolated TSH deficiency Phenotype: Relatively mild symptoms of hypothyroidism due to basal function of thyroid gland. Thyroid gland normal or hypoplastic. T4 and T3 (–), basal TSH (–), TRH-stimulated TSH (–), Prl (+/N). Comments: Can be caused by hypophysitis, empty sella or Langerhans cell histiocytosis, or by mutations of TSH or TRHR. 6 Known genetic defect (TSH-beta, TRHR) Phenotype: Congenital secondary hypothyroidism may not be detected in neonatal screening in many countries, because TSH levels are not increased. T4 and T3 (–), basal TSH (–), TRH-stimulated TSH (–), Prl (N or –). Comment: Different mutations in the coding region of the TSH-beta subunit gene and TRHR. 7 TSH deficiency in combination with other pituitary deficiencies Synonym: Hypopituitarism, combined pituitary hormone deficiency (CPHD). Phenotype: Hypoplastic or normal thyroid, symptoms of CH usually mild. Thyroid hormones (–), TSH (–), Tg (–), GH (–), Prl (–) or gonadotrophins (–). 8 Secondary (pituitary) hypothyroidism Phenotype: Due to pituitary TSH deficiency. TSH (–), T4 (–). Symptoms of hypothyroidism. Variable phenotype depending on other pituitary hormones affected. Comments: Mutations in POU1F1 or PROP1 and TRH receptor gene are coded elsewhere. Malformation syndromes (septo-optic dysplasia, midline defects) and mass lesions may affect pituitary TSH secretion. 9 Tertiary (hypothalamic) hypothyroidism Phenotype: Hypothyroidism due to hypothalamic TRH deficiency. Mostly other hypothalamic hormones are affected as well. TSH (N/–), T4 (–), prolonged TSH rise after TRH injection. 10 Acquired hypothalamic-pituitary hypothyroidism Phenotype: Consequence of pituitary surgery or lymphocytic adenohypophysitis. May occur in very low birth weight infants or critically ill children (non-thyroidal illness syndrome). 11 Isolated hypogonadotrophic hypogonadism and anosmia (Kallman syndrome) Phenotype: Clinically and genetically heterogeneous disorder with a wide spectrum of reproductive function and anosmia. Absence or severely delayed or slowly progressive or abortive puberty. ESPE Classification of Paediatric Endocrine Diagnoses 36 Histologically immature gonads. LH (–), FSH (–), T (–), inhibin-B (–). Hypoplasia of the bulbus olfactorius in MRI. In severe forms hypoplastic genitalia. Comments: Lack of GnRH neurones. X-linked mutations in KAL-1 gene (encodes anosmin-1, a protein involved in olfactory and neuronal migration). Autosomal-dominant loss of function mutations in FGFR-1 gene. Anosmia occurs in all KAL-1 mutations but only a small percentage of FGFR-1 mutations. 12 Isolated hypogonadotrophic hypogonadism and adrenal hypoplasia congenita (DAX1 mutation) Phenotype: X-linked congenital adrenal insufficiency in neonatal period, lack of pubertal development, abnormalities in spermatogenesis. Variable clinical presentation. In female carriers delayed puberty. Hypoplastic adrenals; inadequate testis development, decreased spermatogonia. LH (–), FSH (–), T (–), especially at the time of puberty. Glucocorticoid and mineralocorticoid deficiency. Comments: Deletion or mutation of the NROB1 (DAX1) gene, encoding DAX1, an orphan nuclear receptor. DAX1 expression has been shown in regions of the hypothalamic/pituitary/adrenal/gonadal axis. 13 KAL2 (FGFR1 defect) Comment: Together with KAL mutations, FGR1 mutations are responsible for 20% of cases with Kallmann syndrome. 14 KAL3 and KAL4 Comments: PROKR and PROK2 defects may be responsible for 10% of cases with Kallmann syndrome. 15 Inactivating mutation of GnRHR Phenotype: Lack of pubertal development; wide spectrum of phenotypes. Patients are normosmic. LH (–), FSH (–), T (–). Comment: Autosomal recessive. 16 GPR54 mutation Phenotype: Lack of pubertal development and fertility. LH (–), FSH (–), sex steroids (–). Comments: GPR54 is a G protein-coupled receptor gene, expressed by GnRH neurons. Autosomal recessive. Patients respond to GnRH or Gn treatment. 17 KISS mutation Phenotype: Lack of pubertal development, infertility. Comment: Kisspeptins stimulate gonadotrophins by stimulating GnRH after activation of GPR54. 18 Prohormone convertase 1 mutation Phenotype: Lack of pubertal development. LH (–), FSH (–), sex steroids (–). Comment: Defects in prohormone processing. Pituitary, Hypothalamus, Central Nervous System (CNS) 37 19 Leptin 1 mutation Phenotype: Morbid obesity and hypogonadism. LH (–), FSH (–), sex steroids (–). Comments: Sympathetic system dysfunction, thyroid dysfunction, type 2 DM and immune dysfunction may occur. Increased mortality in obese subjects. 20 Isolated LH deficiency Synonym: Fertile eunuch. Phenotype: Decreased virilisation and fertility. LH (–), T (–). 21 Isolated FSH deficiency Phenotype: Decreased fertility. Decreased number of germ cells. FSH (–). 22 Hypogonadotrophic hypogonadism in combination with other pituitary deficiencies Synonym: Combined pituitary hormone deficiency (CPHD). 23 Known genetic defect, e.g. PROP1, LHX3, HESX1 Phenotype: Mutations in pituitary transcription factors result in combined pituitary hormone deficiency. In LHX3 mutation combination with rigid cervical spine. LH (–), FSH (–), sex steroids (–); depending on the defect combination with other pituitary hormone deficits. PROP1: mutations in the PROP gene are associated with a highly variable phenotype. ACTH deficiency can occur late. HESX1: inactivating mutations in this gene were found in few patients with septo-optic dysplasia. LHX3: mutations in the LHX3 gene which encodes LIM transcription factors responsible for pituitary and nervous system development. 24 Isolated prolactin deficiency Phenotype: Pituitary Prl is synthesised by lactotrophs and mammosomatotrophs which release also GH. Prl is also synthesised in the decidua of the pregnant uterus. In adults, Prl is primarily involved with reproductive functions. In children no symptoms of isolated Prl deficiency. Prl (–). Comments: Predominant negative hypothalamic control by dopamine. The main releasing factor is TRH. 25 POU1F1 mutation Phenotype: Symptoms related to congenital hypothyroidism and GH deficiency. Prl deficiency itself has no clinical sequelae in children. GH (–). Basal and TRH-stimulated TSH (–). Prl (–). Variable phenotype. Comments: AD or AR. POU1F1 is expressed in lactotrophs, somatotrophs and thyrotrophs in which it is involved in the transcription of the GH, Prl and TSHbeta genes. 26 PROP1 mutation Phenotype: Short stature due to complete GH deficiency, congenital hypothyroidism, hypogonadism. Evolving ACTH deficiency in older patients (hyponatraemia, hypoglycaemia). Deficiency of anterior pituitary hormones. Progressive shrinking of the pituitary gland. Occasional pituitary enlargement imposing as adenoma, which can resolve spontaneously. Comments: PROP1 mutations were found in approximately 1/3 of patients with familial combined pituitary hormone deficiency; rare in sporadic cases. ESPE Classification of Paediatric Endocrine Diagnoses 38 27 TSH-producing adenoma Phenotype: TSH- (and Prl-) producing micro- and macroadenomas cause symptoms of hyperthyroidism (and hyperprolactinaemia). TSH (+), T4 (+), Prl (+). Comments: In most cases other pituitary hormones are involved as well. Carcinomas very rare. 28 Gonadotrophin-producing adenoma Phenotype: Functioning adenomas exhibit mainly FSH hypersecretion and ovarian overstimulation. FSH (+), E2 (+). Clinical symptoms of mass lesion (optic chiasm compression, deficiency of other pituitary hormones) may occur. LH-secreting adenoma may cause precocious puberty. LH (+), T (+). Comments: The majority of clinically nonfunctioning pituitary macroadenomas are of gonadotroph origin, LH and FSH are rarely increased. 29 Prolactinoma Phenotype: Prolactinomas may present with delayed puberty. Mostly girls affected: menstrual disorders and galactorrhoea. In large adenomas symptoms of intracranial mass lesion. Prl (+), pulsatile secretion, variable response to TRH stimulation. Comments: 40% of acromegalic patients have hyperprolactinaemia. The extent of Prl elevation may discriminate between nonfunctioning macroadenoma and macroprolactinoma. 30 Hyperprolactinaemia of other cause Phenotype: Pituitary stalk compression by a tumour may lead to hyperprolactinaemia by decreasing Prl inhibition by dopamine. In primary hypothyroidism the serum level of TRH, which is the primary Prl-releasing factor, is increased. 31 Septo-optic dysplasia (De Morsier syndrome) Phenotype: Growth retardation, visual impairment, nystagmus; hypothalamic dysfunction and pituitary failure may occur. Neonatal hypoglycaemia and seizures. Developmental anomalies of the midline structures of the brain like hypoplasia of optic nerves, agenesis of septum pellucidum and agenesis of corpus callosum. Variable pituitary hormone deficiencies. Very variable phenotype. Comment: HESX1 mutations were found only in few cases. 32 Other midline defects: cleft palate, central maxillary incisor syndrome, EEC syndrome (ectodactyly-ectodermal dysplasia-clefting syndrome) Phenotype: Developmental anomalies of the midline may affect the pituitary to a variable extent, mostly GH deficiency. Comments: Common development of palate/Rathke pouch/pituitary. The central incisor syndrome may be associated with holoprosencephaly and various other congenital anomalies. 33 Ectopic neurohypophysis, absent infundibulum and hypoplastic adenohypophysis Phenotype: Short stature due to GH deficiency and other symptoms due to TSH, ACTH and gonadotrophin deficiency, mostly panhypopituitarism. MRI shows hypoplastic adenohypophysis and small sella, hypoplastic or absent infundibulum and ectopic neurohypophysis at the median eminence. Basal and stimulated GH (–), TSH (–), Gn (–), ACTH (–). Comments: This condition is likely to be the end result of perinatal disruption of the hypophyseal portal system. The ectopic neurohypophysis functions normally. Pituitary, Hypothalamus, Central Nervous System (CNS) 39 34 Hamartoma Phenotype: Precocious puberty of central type, psychomotor delay, seizures. Benign tumours composed of neurons, astrocytes and oligodendroncytes; appears attached to the tuber cinereum or the floor of the 3rd ventricle. The tumour may express GnRH. 35 Chromosomal disorders Phenotype: Turner syndrome, 18p deletion syndrome, XXXXY syndrome. Brain malformations and pituitary hormone deficiencies (mainly GH deficiency) were found in a variable degree. 36 Chromosomal instability syndromes Phenotype: Fanconi syndrome, Bloom syndrome, Nijmegen breakage syndrome. GH deficiency has been described. GH treatment in these cases may cause increased susceptibility to malignancies. 37 Empty sella syndrome Phenotype: In most cases asymptomatic; rarely clinical manifestation with headache, rhinorrhoea, impaired visual field and benign intracranial hypertension. Rarely with pituitary deficiencies. GH (–), TSH (–), LH (–), FSH (–), Prl (+). Comments: Disorder of the anlage of the diaphragm of the sella. Enlarged pituitary fossa by herniated arachnoid content. In unselected autopsies up to 20%. 38 Craniopharyngioma Phenotype: Symptoms depend on the relationship to the pituitary, hypothalamus or optic chiasma. Hypopituitarism by compression of the adenohypophysis. Compression of the hypophyseal stalk causes hyperprolactinaemia. Visual field disturbance, intracranial pressure, headache, vomiting, papilloedema, somnolence. After surgery virtually all patients show hypopituitarism. Derives from remnant of Rathke pouch, supra- or intrasellar, calcifications. Comment: Approximately 10% of childhood tumours. 39 Nonfunctional pituitary adenomas Phenotype: A mass lesion impinging on the pituitary or hypothalamus may cause local neurological effects on the optic tract, the cavernous sinus, temporal and frontal lobe and may cause symptoms of increased intracranial pressure. Involvement of the hypothalamus (temperature dysregulation, appetite and thirst disorders, etc.) and pituitary (hormone deficiencies). 40 Other benign structures Phenotype: May cause the same phenotype as in non-functional pituitary adenomas (6F.1a.2). 41 Isolated glioma Phenotype: Brain tumours originating from neuroglia. Symptoms depend on impingement on local structures (chiasmatic/hypothalamic area), hydrocephalus. Variable pituitary hormone deficits. 42 Germinoma, dysgerminoma Phenotype: Germinomas arise near the base of the hypothalamus and affect vasopressin axons thereby causing neurohormonal diabetes insipidus. Can be very small (pituitary stalk thickening) and undetectable for several years following the onset of polyuria. hCG (+), hCG in CSF, diabetes insipidus. Dysgerminoma may produce gonadotrophins and give rise to precocious puberty. ESPE Classification of Paediatric Endocrine Diagnoses 40 43 Leukaemia, lymphoma Phenotype: Most frequent causes of CNS-affecting malignant diseases. May affect hypothalamicpituitary axis by leukaemic infiltration (leukaemic meningitis) or as a consequence of treatment. Comments: Normal growth may be maintained despite biochemical GH deficiency due to hypothalamic hyperphagia in children with hypothalamic disorder. 44 Pinealoma Phenotype: Pineal parenchymal tumours (pineocytoma, pineoblastoma) or pure pineal germinomas (occur mainly in boys). Symptoms of mass lesion, obstructive hypertension. Melatonin has no diagnostic significance. 45 Isolated glioma Phenotype: Tumours outside the hypothalamo-pituitary region may cause symptoms of mass lesion and obstructive hypertension and thereby affect pituitary hormone secretion. 46 Medulloblastoma Phenotype: Embryonal tumour of the cerebellum, metastases into the CSF. Most common malignant brain tumour in children. Affection of the hypothalamus-pituitary axis as consequence of treatment (surgery, high dose irradiation, chemotherapy). Comment: Mutations of genes encoding proteins involved in embryonic growth factors or mutations of growth factor receptors are frequently found. 47 Langerhans cell histiocytosis (LCH) Phenotype: Can occur as an isolated lesion, as multifocal or as widespread systemic disease. Focal proliferation/accumulation of Langerhans cells. Class I, II and III histiocytosis based on histopathologic findings. Single or multiple lesions of the bone, skin involvement, exophthalmos, pulmonary infiltrates, pituitary involvement or hypothalamic dysfunction. Diabetes insipidus in approximately 25%, variable deficiencies of the anterior pituitary. LCH has an extremely variable clinical presentation. 48 Systemic lupus erythematodes (SLE) Phenotype: SLE is a multisystem autoimmune disorder which can present with neurological complications. Brain lesions on MRI. The pituitary-thyroid and pituitary-gonadal axis may be affected in newly diagnosed, untreated patients. Comments: Genetic predisposition, female preference. 49 Neurosarcoidosis Phenotype: Infiltrative granulomatous disease which may involve hypothalamic-pituitary area. Central diabetes insipidus, hypogonadism, GH deficiency, hyperprolactinaemia. Thickening of the pituitary stalk. Difficult differentiation from lymphocytic hypophysitis (biopsy). 50 Lymphocytic neurohypophysitis Phenotype: Recent pregnancy or other autoimmune disease in 50% of cases, occasionally after pituitary surgery. Diffuse destruction of pituitary and infundibulum. Affection of anterior and posterior pituitary hormone axis. Central diabetes insipidus, LH (–), FSH (–), T/E2 (–), TSH (–), T4 (–), ACTH (–), cortisol (–), GH (–). Pituitary, Hypothalamus, Central Nervous System (CNS) 41 51 Haemochromatosis Phenotype: May lead to pituitary infiltration, deposits in pituitary cells. Idiopathic or secondary (thalassaemia or sickle cell disease). Correlation with serum ferritin levels; MRI shows decreased signal intensity. Mainly gonadotrophic axis affected, delayed puberty. LH (–), FSH (–), T or E2 (–). 52 Meningitis Phenotype: Viral or bacterial meningoencephalitis can be associated with anterior and posterior pituitary insufficiency which may resolve after cure of the disease. Central diabetes insipidus is frequently seen in congenital meningitis of various aetiology. 53 Abscess of pituitary Phenotype: Intrasellar infection presenting with headache, visual changes, fever, meningismus and endocrine abnormalities. Imaging demonstrates pituitary mass. Correct diagnosis mostly only after (surgical) treatment. Generally pituitary dysfunction does not recover after treatment. 54 Congenital infection Phenotype: Prenatal rubella infection in the first trimester causes dysmorphic stigmata (microphthalmos, microcephaly), cardiac anomalies, dystrophia, GH deficiency has been described. Hypothyroidism was described in congenital immunodeficiency syndromes. Neurohormonal diabetes insipidus was found in congenital cytomegalovirus infection. GH deficiency was found in Schwachmann-Diamond syndrome (pancreatic insufficiency, recurrent infections, metaphyseal anomalies). 55 CNS surgery Phenotype: Tumours in the sella or hypothalamus region are mostly associated with hypothalamic-pituitary hormone deficits; after surgery deficits will usually get worse, rarely recovery. Usually multiple pituitary hormone deficits. Brain surgery outside the hypothalamo-pituitary region may affect hormone production and secretion by pressure, blood circulatory disturbance or haemorrhage. Trans-sphenoidal surgery frequently followed by SIADH (hyponatraemia, elevated vasopressin). 56 Head trauma (traumatic brain injury, TBI) Phenotype: Up to 25–50% of patients have been reported to develop anterior pituitary deficiency, in particular GH deficiency. Can evolve over years. Systematic screening of pituitary function in all patients after head trauma and TBI appears warranted. 57 Hypoxic injury Phenotype: Hypoxic brain injury, especially in the neonate, can produce irreversible tissue damage. Predominantly diabetes insipidus as a sign of severe brain damage. Anterior pituitary hormone deficits may develop with some delay. 58 Irradiation Phenotype: Cranial irradiation for prophylaxis or treatment of leukaemic meningitis or for treatment of various brain tumours. Radiotherapy has a dose-dependent effect on hypothalamic-pituitary hormone secretion, GH secreting cells being most sensitive. Hormone deficits occur years after treatment. Comment: May also affect cartilage plates and bone growth. ESPE Classification of Paediatric Endocrine Diagnoses 42 59 Drugs, e.g. chemotherapy, alcohol Phenotype: Chemotherapy affects growth mainly by a catabolic condition and by direct effects on bone, possible effects on hypothalamus. Foetal alcohol syndrome results from maternal alcohol use during pregnancy (facial abnormalities, impairment in neurodevelopment and growth). Cases with GH deficiency have been reported. 60 Anorexia nervosa Phenotype: Amenorrhoea, weight loss, behavioural changes (fear of gaining weight, disturbance of self-evaluation of body shape, hyperactivity, sleep disturbance). Endocrine abnormalities represent an adaptation to starvation. LH (–), FSH (–), E2 (–), T4 (–), TSH (N), Prl (N), basal GH (+), IGF1 (–). Generally in young women under age 25. Female:male ratio 9:1. 61 Emotional deprivation (psychosocial dwarfism, deprivational dwarfism) Phenotype: Short stature, abnormal behaviour patterns (perverted or voracious appetite, enuresis, encopresis, insomnia), occurs in children with a disturbed social environment. GH (–), IGF1 (–), occasional TSH and ACTH deficiency. Reversible if placed in a favourable psychosocial environment. Pituitary, Hypothalamus, Central Nervous System (CNS) 43 ESPE Code Diagnosis 7 THYRO I D D I SO R D E R S 7A HYPOTHYROIDISM1 7A.1 Congenital (primary) hypothyroidism (permanent)2 Included: Late-onset hypothyroidism due to thyroid dysgenesis or dyshormonogenesis Disorders classified elsewhere: Secondary and tertiary hypothyroidism (6A.2) Thyroid hormone resistance (7E.3) Developmental defects/thyroid dysgenesis3 Athyrosis/agenesis4 Hypoplasia/hypogenesis5 Ectopy6 Inherited defects of thyroid hormone biosynthesis/thyroid dyshormonogenesis7 Iodine transport defect8 Organification defects due to an abnormality in the TPO enzyme or in the H2O2 generating system (includes Pendred syndrome)9 7A.1a 7A.1b 7A.1b.1 7A.1b.2 7A.1b.3 7A.1c 7A.1c.1 7A.1c.2 OMIM ICD10 E03.1 #218700 E03.1 #218700 E03.1 #218700 Q89.2 E07.1 #274400 #274500 #274600 #607200 7A.1c.3 7A.1c.4 7A.1d Defective thyroglobulin synthesis or transport10 Abnormal iodotyrosine deiodinase (dehalogenase) activity11 TSH-receptor defects/TSH unresponsiveness12 +188450 %274800 #218700 E07.9 #275200 7A.1y 7A.1z Other specified disorders Other disorders, unspecified 7A.2b.1 Transient congenital hypothyroidism Iodine deficiency13 Drug induced Maternal anti-thyroid drug therapy14 7A.2b.2 Iodine excess due to iodinated contrast media 7A.2b.3 Iodine excess from other sources15 7A.2b.8 Other specified goitrogen 7A.2b.9 Other goitrogen, unspecified 7A.2c Maternal thyroid autoantibodies16 7A.2 7A.2a 7A.2b E00 P72.2 E03.2 P72.2 E03.2 P72.2 E03.2 P72.2 E03.2 P72.2 E03.2 ESPE Classification of Paediatric Endocrine Diagnoses P72.2 44 ESPE Code Diagnosis 7A.2d 7A.2y 7A.2z Thyroid dysfunction in prematurity (including transient hypothyroxinaemia, transient primary hypothyroidism, transient hyperthyrotropinaemia, low T3 syndrome) Other specified disorder Idiopathic 7A.3z Acquired primary hypothyroidism Due to disorder classified elsewhere: Non-autoimmune thyroiditis (7E.4) Autoimmune thyroiditis/Hashimoto‘s thyroiditis with decreased thyroid function17 Iodine deficiency18 Iatrogenic19 Post-irradiation Post-operative Iodine excess20 Drug-induced, including environmental conditions (goitrogens)21 Systemic diseases22 Other specified disorders Idiopathic 7B H Y P E R T H Y R O I D I S M 23 7B.1 Congenital hyperthyroidism (neonatal thyrotoxicosis)24 Disorders classified elsewhere: Isolated pituitary resistance to thyroid hormone (7E.3b) Maternal autoimmune thyroid disease25 (neonatal Graves’ disease) (usually transient) Activating TSH-receptor mutation26 G-protein mutations27 As part of McCune Albright [primary 14B.22] Other 7A.3 7A.3a 7A.3b 7A.3c 7A.3d 7A.3d.1 7A.3d.2 7A.3e 7A.3f 7A.3g 7A.3y 7B.1a 7B.1b 7B.1c 7B.1d 7B.1d.1 7B.1d.2 7B.2 7B.2a 7B.2b 7B.2c OMIM Acquired primary hyperthyroidism Due to disorder classified elsewhere: Thyroid adenoma (7D.1) Hyperfunctioning carcinoma (7D.2) Non-autoimmune thyroiditis (7E.4) Steinert syndrome (14B.35) Graves’ disease, hashitoxicosis (Hashimoto’s toxicosis)28 Exogenous Thyroid Disorders ICD10 P72.2 P72.2 P72.2 %140300 E06.3 E01 E89.0 E03.2 E03.2 E03.8 E03.8 E03.9 P72.1 #609152 E05.8 E05.8 #174800 E05.8 E05.8 %140300 E05.0 45 ESPE Code Diagnosis OMIM 7B.2z Excessive intake of thyroid hormones29 Iodine induced (‘Jod-Basedow’) Other specified disorder, e.g. choriocarcinoma, hydatidiform mole Idiopathic 7C GOITRE 7C.0 Due to disorder classified elsewhere Supplementary code only If hypothyroid use code from 7A If hyperthyroid use code from 7B 7C.1 Euthyroid goitre Disorders classified elsewhere: Non-autoimmune thyroiditis (7E.4) Dyshormonogenetic Iodine transport defect30 #274400 Organification defects due to an abnormality in the TPO enzyme #274500 or in the H2O2 generating system (includes Pendred syndrome)31 #274600 7B.2c.1 7B.2c.2 7B.2y 7C.1a 7C.1b 7C.1b.1 7C.1b.2 ICD10 E05.4 E05.4 E05.9 E05.9 E04 E07.1 #607200 7C.1b.3 7C.1b.4 7C.1c Defective thyroglobulin synthesis or transport32 Abnormal iodotyrosine deiodinase (dehalogenase) activity33 TSH-receptor defects/TSH unresponsiveness +188450 %274800 #218700 E07.9 #275200 7C.1z Iodine deficiency34 Autoimmune/Hashimoto’s thyroiditis with normal thyroid function35 Drug-induced/goitrogen exposure Nodular goitre due to cysts or haemorrhage36 Other specified disorder Idiopathic (juvenile goitre, adolescent goitre, simple goitre)37 7D THYROID TUMOURS 7D.1 Adenomas38 7D.2 Carcinomas Papillary39 Follicular40 7C.1d 7C.1e 7C.1f 7C.1g 7C.1y 7D.2a 7D.2b ESPE Classification of Paediatric Endocrine Diagnoses E01, E02 %140300 E06.3 E04.8 E04.8 E04.9 D34 C73, D44 #188550 46 ESPE Code Diagnosis OMIM ICD10 7D.2c.2 Medullary (C cell carcinoma)41 Sporadic Part of MEN 2A [primary 14C.5b] #171400 D44.8 7D.2c.3 MEN 2B [primary 14C.5c] #162300 7D.2c.4 Other familial forms (RET, NTRK1) Undifferentiated/anaplastic Other specified carcinoma Other carcinoma, unspecified #155240 7D.2c 7D.2c.1 M8360/1 D44.8 M8360/1 7D.2d 7D.2y 7D.2z 7D.8 7D.9 Other specified tumour E.g. lymphoma, sarcoma D44 Other tumour, unspecified D44.0, C73 C73 D34 7E OTHER THYROID DISORDERS 7E.1 Sick-euthyroid syndrome Excluded: Thyroid dysfunction in prematurity (7A.2d) 7E.2 Disorders of thyroid hormone transport42 TBG deficiency TBG excess Transthyretin (TTR) variants Familial dysalbuminaemic hyperthyroidism 7E.2a 7E.2b 7E.2c 7E.2d 7E.3 7E.3a 7E.3b 7E.3c Thyroid hormone resistance syndromes43 Generalised thyroid hormone resistance (GTHR) Pituitary resistance to thyroid hormone (PitRTH) Peripheral resistance to thyroid hormone (PRTH) 7E.4c Non-autoimmune thyroiditis Non-autoimmune thyroiditis due to viral or bacterial agents Acute suppurative thyroiditis44 Subacute thyroiditis 7E.5 Other thyroid disorders, unspecified 7E.4 7E.4a 7E.4b Thyroid Disorders E07.8 E07.8 +314200 +176300 +176300 E07.9 #188570 #145650 #188570 E06.0 E06.0 E06.1 47 1 Hypothyroidism Phenotype: Decreased activity, dry skin and hair, excessive weight gain secondary to fluid retention, growth failure, retarded bone age. TSH (+ in primary; –/N in secondary/tertiary hypothyroidism), FT4 (–), FT3 (–). 2 Congenital (primary) hypothyroidism (CH) Phenotype: Delayed skeletal maturation, enlarged fontanels, constipation, enlarged tongue, lethargy, developmental delay. Thyroid gland either absent, hypoplastic, ectopic, normally-sized or enlarged. In all forms of primary hypothyroidism: low thyroid hormones, elevated TSH. Comments: Heterogenous pathogenesis. 80% due to developmental defects. Neonatal screening enables early diagnosis and treatment. 3 Developmental defects/thyroid dysgenesis Phenotype: Different forms: athyrosis, ectopy, hypoplasia. Diagnosis by thyroid imaging (ultrasound, scintigraphy). 4 Athyrosis/agenesis Phenotype: Most pronounced form of hypothyroidism. Remnants of fibrotic tissue might be detectable. 5 Hypoplasia/hypogenesis Phenotype: Similar as in athyrosis. Remnants of normal tissue can be visualised, but of smaller size than normal. 6 Ectopy Phenotype: Hypothyroidism usually milder than in athyrosis. Normal tissue can be visualised in an abnormal location. 7 Inherited defects of thyroid hormone biosynthesis/thyroid dyshormonogenesis Phenotype: Symptoms and signs of hypothyroidism. Goitre is rare in the neonate, but frequent in older children. 8 Iodine transport defect Phenotype: Inability of the thyroid to maintain a concentration difference of readily exchangeable iodine between the plasma and the thyroid gland. The defect is also found in the salivary gland and gastric mucosa. It is presumed to arise either because of a deficient supply of energy for the transport system or because of abnormality of a carrier or receptor substance. Failure to concentrate radioiodide by the salivary gland (saliva/plasma (123) I ratio was 1.6, in contrast to the normal ratio of more than 20) and the clinical and biologic response to potassium iodide treatment. Comment: The defect in iodide transport is produced by mutations in the sodium-iodide symporter gene (SLC5A5). 9 Organification defect Phenotype: Normal or enlarged gland in ultrasound, rapid uptake of radioactive iodine, but also rapid discharge after perchlorate administration (perchlorate discharge test). ESPE Classification of Paediatric Endocrine Diagnoses 48 Comment: Defect of iodine organification (thyroidperoxidase and H2O2 generation defects have been identified). 10 Defective thyroglobulin synthesis or transport Phenotype: Normal or enlarged gland in ultrasound, rapid uptake of radioactive iodine, normal discharge after perchlorate administration (perchlorate discharge test). Tg (–/N/+) (immunoreactivity might be normal). Comment: Defect of the thyroglobulin molecule, which results in an ineffective thyroid hormone production. 11 Abnormal iodotyrosine deiodinase (dehalogenase) activity Phenotype: Normal or enlarged gland in ultrasound, symptoms of CH usually milder than in athyrosis. MIT and DIT elevated in serum and urine. Comment: Defect of the deiodinase which leads to failure of deiodination of iodotyrosine and loss of iodine in the form of MIT and DIT in the urine, frequent development of goitre. 12 TSH-receptor defects/TSH unresponsiveness Phenotype: Normal or hypoplastic thyroid, symptoms of CH usually varying from severe to mild. Tg (N/–). Comment: Defect of the TSH receptor may be associated with euthyroid hyperthyrotropinaemia or hypothyroidism. 13 Iodine deficiency Synonym: Endemic cretinism. Phenotype: Enlarged, normal or hypoplastic thyroids, symptoms of CH varying from severe to mild. Tg (+), urinary iodine excretion (–). Comments: Congenital hypothyroidism due to iodine deficiency is common in developing countries. It is preventable by iodine prophylaxis. In some industrialised countries with lesser degrees of iodine deficiency, the incidence of congenital hypothyroidism and hyperthyrotropinaemia is increased as well. 14 Maternal anti-thyroid drug therapy Phenotype: Normal or enlarged thyroid, mild symptoms of CH. Comments: Transplacental passage of anti-thyroid drugs may result in the inhibition of the foetal thyroid function. Condition is transient and disappears with elimination of the anti-thyroid drug from the infant’s circulation. 15 Iodine excess Phenotype: Enlarged or normal thyroids, symptoms of CH usually mild, however, long-term sequelae are possible. Tg (+), extremely elevated urinary iodine excretion. Comments: Administration of large amounts of iodide, e.g. disinfectants or contrast media to the mother or infant leads to transient suppression of thyroid function (Wolff-Chaikoff effect). Transient hypothyroidism due to iodine excess is frequent in newborns and premature infants in intensive care. Thyroid Disorders 49 16 Maternal thyroid autoantibodies Phenotype: Normal or hypoplastic thyroid, symptoms of CH varying from severe to mild. Anti-thyroid antibodies detectable. Comments: Transplacental passage of thyroid autoantibodies (thyroid peroxidase (TPO), TSH receptor, cytotoxic) may disturb thyroid function and development of the foetal thyroid gland. Usually transient condition, in some cases with cytotoxic and thyroid blocking IgG it is permanent. 17 Autoimmune thyroiditis/Hashimoto’s thyroiditis with decreased thyroid function Phenotype: Thyroid enlarged or normally sized, abnormal echo pattern in ultrasound investigation, mild to severe symptoms of hypothyroidism, short stature. Histology: inflammatory signs, lymphocytic infiltration. Comments: Hypothyroidism due to an inflammation and destruction of thyroid caused by anti-thyroid autoantibodies or viral/bacterial agents. Incidence variable, more frequent in iodine replete areas. 18 Iodine deficiency Phenotype: Enlarged or normal thyroids, symptoms of CH usually mild, short stature is the most prominent feature in infants and children. Tg (+), low urinary iodine excretion. Comment: Hypothyroidism due to iodine deficiency caused by inadequate nutritional supply or substances which inhibit iodine uptake, e.g. soy, cassava. 19 Iatrogenic Phenotype: Absent or diminished thyroid tissue, symptoms of hypothyroidism. Comment: Hypothyroidism following the surgical or radiotherapy treatment of hyperthyroidism, cancer or nodular goitre. 20 Iodine excess Phenotype: Enlarged or normal thyroid, symptoms of CH usually mild. Tg (+), extremely elevated urinary iodine excretion. Comment: Administration of large amounts of iodide, e.g. disinfectants or contrast media, infrequently leads to transient suppression of thyroid function in an older child (Wolff-Chaikoff effect). 21 Drug-induced Phenotype: Thyroid enlarged or normally sized, mild to severe symptoms of hypothyroidism, short stature. Comment: Hypothyroidism due to administration of drugs which block thyroid function (perchlorate, carbimazole, methimazole) or iodine uptake (perchlorate, amiodarone) or cause autoimmune inflammation (interferon). 22 Systemic diseases Phenotype: Hypothyroidism, storage of iron, cystin, etc., in the thyroid, usually small glands. Comment: Hypothyroidism as a symptom of systemic disease (e.g. cystinosis, -thalassemia), usually caused by storage of non-thyroidal material or inflammation in the thyroid. ESPE Classification of Paediatric Endocrine Diagnoses 50 23 Hyperthyroidism (thyrotoxicosis) Phenotype: Rapid growth associated with restlessness, tremor, heat intolerance, diarrhea, polydipsia and poor concentration. T4 (+), FT4 (+), T3 (+), TSH (–). TSH suppressed despite TRH stimulation. 24 Congenital hyperthyroidism (neonatal thyrotoxicosis) Phenotype: Manifestations of hyperthyroidism before birth, and in neonatal period. 25 Neonatal thyrotoxicosis due to maternal autoimmune thyroid disease Synonym: Neonatal Graves’ disease. Phenotype: In utero tachycardia can occur. From birth (or a few days later): thyroid enlarged or normally sized, abnormal echo pattern in ultrasound investigation, mild to severe symptoms of hyperthyroidism, failure to thrive, tachycardia, exophthalmos, restlessness. T4 (+), T3 (+), TSH (–), measurable stimulating auto-antibodies. Mother has circulating thyroid-stimulating immunoglobulins (TSI). Comments: Hyperthyroidism due to the transplacental passage of thyroid autoantibodies which stimulate the thyroid gland, usually TSH receptor antibodies (TRAb) (TSI). Pregnant women with a history of thyroid disease should be screened for TSI in early pregnancy. In case of positive TSI, foetal heart rate has to be checked regularly. Increased familial occurrence. More frequent in iodinereplete areas. 26 Activating TSH-receptor mutation Phenotype: Thyroid normally sized, rarely enlarged at birth, normal echo pattern in ultrasound investigation, severe symptoms of hyperthyroidism, failure to thrive, tachycardia, relatively resistant to anti-thyroid drug treatment. T4 (+), T3 (+), TSH (–), no measurable auto-antibodies. Comment: Hyperthyroidism due to constitutively activating mutations of the TSH receptor. 27 G-protein mutations Phenotype: Thyroid normally sized, rarely enlarged, normal echo pattern in ultrasound investigation, severe symptoms of hyperthyroidism, failure to thrive, tachycardia, relatively resistant to antithyroid drug treatment. T4 (+), T3 (+), TSH (–), no measurable auto-antibodies. Comment: Hyperthyroidism due to constitutively activating mutations of the G-protein signalling pathway, sometimes combined with dysfunction of other endocrine cells, e.g. precocious pseudopuberty, hypo- or hyperparathyroidism (McCune-Albright). 28 Graves’ disease Synonym: Hashitoxicosis. Phenotype: Thyroid enlarged, abnormal echo pattern in ultrasound investigation, mild to severe symptoms of hyperthyroidism, tachycardia, weight loss, nervousness, increased blood pressure, exophthalmos, most severe symptoms evident in thyroid storm. T4 (+), T3 (+), TSH (–), measurable stimulating auto-antibodies (anti-TSH receptor (TRab) and anti-TPO, and anti-thyroglobulin (Tg) autoantibodies). Comment: Increased familial occurrence. 29 Excessive intake of thyroid hormones Phenotype: Thyroid not enlarged, severe symptoms of hyperthyroidism, tachycardia, weight loss, nervousness, increased blood pressure, nausea. T4 (+), T3 (+), TSH (–). Thyroid Disorders 51 Comments: Hyperthyroidism due to large doses of exogenous thyroid hormones. Doses of mg to g of L-thyroxine are needed to produce symptoms, while T3 is more toxic with symptoms already in μg amounts. 30 Iodine transport defect Phenotype: Goitre, enlarged gland in ultrasound, but not visualised in scintigraphy. Tissue can be visualized by decreased iodine content of saliva. T4 (N), TSH (N/+). Comment: Defect of iodine transport into the thyroid and salivary glands. 31 Organification defect Phenotype: Goitre, enlarged gland in ultrasound, rapid uptake of radioactive iodine, but also rapid discharge after perchlorate administration (perchlorate discharge test). T4 (N/–), T3 (N), TSH (N/+). Comment: Defect of iodine organification (thyroid peroxidase and H2O2 generation defects have been identified). 32 Defective thyroglobulin synthesis or transport Phenotype: Goitre, enlarged gland in ultrasound, rapid uptake of radioactive iodine, normal discharge after perchlorate administration (perchlorate discharge test). T4 (N), TSH (N), Tg (–/N/+) (immunoreactivity might be normal). Comment: Defect of the thyroglobulin molecule, which results in an ineffective thyroid hormone production. 33 Abnormal iodotyrosine deiodinase (dehalogenase) activity Phenotype: Goitre, enlarged gland in ultrasound. T4 (N), TSH (N), MIT and DIT elevated in serum and urine. Comments: Defect of the deiodinase which leads to failure of deiodination of iodotyrosine and loss of iodine in the form of MIT and DIT in the urine, frequent development of goitre. 34 Iodine deficiency Phenotype: Enlarged thyroid. Increased radioactive iodine uptake. There can be a cold nodule. T4 (N), TSH (N), Tg (+), urinary iodine excretion (–). Comments: Congenital cases: goitre due to iodine deficiency caused by inadequate nutritional supply or substances which inhibit iodine uptake, e.g. soy, cassava. Acquired cases: goitre development due to iodine deficiency is common in many developing countries but also in some industrialised countries with lesser degrees of iodine deficiency. Possible influence of IGF-1, the incidence of juvenile goitre ranges from 0 to 60% in correlation to iodine supply. Can be associated with nodular goitre. 35 Autoimmune/Hashimoto’s thyroiditis with normal thyroid function Phenotype: Thyroid enlarged, abnormal echo pattern in ultrasound investigation. Auto-antibodies (anti-TPO, anti-Tg, rarely anti-TSH receptor) present. T4 (N) and TSH (N) found in euthyroid cases. Comments: In euthyroid cases the compensatory power of the thyroid is sufficient for normal thyroid hormone production, albeit with an increased gland volume. Increased familial occurrence. ESPE Classification of Paediatric Endocrine Diagnoses 52 36 Nodular goitre due to cysts or haemorrhage Phenotype: Goitre, cystic appearance in ultrasound, cold nodule in scintigraphy. T4 (N), TSH (N), no antibodies. Comments: Nodular goitre, nodules represent cysts or haemorrhages. More prevalent in iodine deficient areas. 37 Idiopathic (juvenile goitre, adolescent goitre, simple goitre) Phenotype: Thyroid enlarged. In nodular goitre single or multiple nodules in ultrasound, usually representing cold, very rarely warm or hot nodules in scintigraphy. T4 (N), TSH (N). Comment: Goitre due to multiple nodule formation, probably due to somatic mutations in genes for thyroid specific proteins. 38 Adenomas Phenotype: Thyroid not enlarged, thyroid nodule demonstrable in ultrasound. In cases with hyperthyroidism a hot nodule is found in scintigraphy, and there are mild to severe symptoms of hyperthyroidism, tachycardia, weight loss, nervousness, increased blood pressure. Thyroid function can also be normal. Histology: single or multiple nodules without inflammatory signs or lymphocytic infiltration. Comment: Hyperthyroidism due to a hyperfunctioning thyroid nodule carrying a somatic mutation of the TSH receptor gene or G-proteins causing constitutive activation of the thyroid cells. 39 Papillary carcinoma Phenotype: Single or multiple nodules in ultrasound, usually representing as cold, very rarely as warm nodules in scintigraphy, frequently early lymph node metastases, bilateral occurrence, favourable prognosis. T4 (N), TSH (N). Comments: Most common thyroid tumour in children, relatively differentiated due to rearrangements of ret-proto-oncogenes. More frequent after neck irradiation. 40 Follicular carcinoma Phenotype: Single or multiple nodules in ultrasound, usually representing as cold, very rarely as warm nodules in scintigraphy, infrequently lymph node metastases, bilateral occurrence, vascular invasion common. T4 (N), TSH (N). Comment: Rare, hard to distinguish from follicular adenoma. More frequent after neck irradiation. 41 Medullary (C cell carcinoma) Phenotype: Single or multiple nodules in ultrasound, usually representing as cold nodules in scintigraphy, early metastasis in lungs and lymph nodes. T4 (N), TSH (N). Elevated calcitonin (after pentagastrin). Comments: In 75% sporadic. Rare tumour caused by ret-proto-oncogene mutations, frequently found in MEN IIa and b, which is hard to distinguish from follicular adenoma. In families with MEN or MTC, children should be screened for mutations of the MEN genes. Calcitonin frequently only increased after pentagastrin stimulation. Thyroid Disorders 53 42 Disorders of thyroid hormone transport Phenotype: Normal thyroid, euthyroidism. T4 (–), FT4 (N), TSH (N). Comment: Usually euthyroid conditions with low measurements of total thyroid hormones but normal free thyroid hormones due to defects of thyroid hormone binding proteins (TBG, albumin). 43 Thyroid hormone resistance syndromes Phenotype: Ranging from overt hypothyroidism with deafness and mental retardation to mild hypothyroidism combined with hyperthyroid symptoms, e.g. tachycardia. Normal thyroid. T4 (+), TSH (+). Comment: Hypothyroidism due to defects of the nuclear T3 receptor with a variable clinical picture ranging from overt hypothyroidism to a mixed clinical picture of hypothyroidism and hyperthyroidism due to different regulation of T3 receptors in different tissues. 44 Acute suppurative thyroiditis Phenotype: Goitre, pain, fever, dysphagia (acute) or weakness, malaise, tenderness of the thyroid (sub-acute), elevated ESR and CRP. T4 (N/–/+), TSH (N/+/–), no antibodies. ESPE Classification of Paediatric Endocrine Diagnoses 54 ESPE Code Diagnosis 8 AD R E NAL D ISO R D E R S 8A PRIMARY ADRENAL INSUFFICIENCY 8A.1 Congenital adrenal hyperplasia (CAH) Cholesterol side-chain cleavage deficiency (lipoid CAH) (P450scc)1 3beta-Hydroxysteroid dehydrogenase deficiency2 8A.1a 8A.1b OMIM ICD10 #201710 E25 E25.0 +201810 E25.0 *109715 8A.1c 8A.1c.1 8A.1c.2 8A.1c.3 8A.1d 8A.1e 8A.1e.1 8A.1e.2 8A.1e.3 8A.1f 8A.1g 8A.1h 8A.1z 21-Hydroxylase deficiency (P450c21)3 Salt-wasting 21-OHD4 Simple virlising 21-OHD5 Non-classical 21-OHD6 11beta-Hydroxylase deficiency7 P450c11AS deficiency Corticosterone methyl oxidase deficiency type I8 Corticosterone methyl oxidase deficiency type II9 Glucocorticoid suppressible hypertension10 17alpha-Hydroxylase/17/20 lyase deficiency11 P450 oxidoreductase deficiency (may be part of Antley Bixler congenital malformation syndrome)12 Glucocorticoid receptor defect13 Other disorders, unspecified 8A.2z Other causes of adrenal insufficiency Congenital adrenal hypoplasia DAX-1 (NROB1) mutation14 (also classified as 6A.3a.2 if combined with hypogonadotrophic hypogonadism) Idiopathic Adrenoleukodystrophy (Schilder’s disease) and adrenomyeloneuropathy15 Primary xanthomatosis (Wolman’s disease)16 Familial glucocorticoid deficiency (ACTH insensitivity, hereditary unresponsiveness to ACTH)17 Triple A (Allgrove) syndrome18 Pseudohypoaldosteronism, type 119 Other syndromes, unspecified 8A.3 Autoimmune adrenalitis (Addison’s disease)20 8A.2 8A.2a 8A.2a.1 8A.2a.2 8A.2b 8A.2c 8A.2d 8A.2e 8A.2f Adrenal Disorders +201910 E25.0 #202010 E25.0 *124080 E25.0 E25.0 E25.0 #103900 E25.0 #202110 #201750 E25.0 E25.0 +138040 E25.0 E25.9 #300200 Q89.1 #202370 E71.3 +278000 Q89.1 #202200 Q89.1 #231550 Q89.1 #264350 Q89.1 Q89.1 E27.1 55 ESPE Code Diagnosis 8A.3a 8A.3b 8A.4 8A.4a 8A.4b 8A.4c 8A.4d 8A.4y Infections Tuberculosis21 Fungal infections22 Bacterial sepsis23 AIDS24 Other specified infections 8A.5z 8A.9 Idiopathic 8B SECONDARY ADRENAL INSUFFICIENCY 8A.5a 8A.5y ICD10 Disorders classified elsewhere: Part of autoimmune polyglandular syndromes: – Type 1 classified as 14C.4a – Type 2 classified as 14C.4b Other types (see 14C.4c, d and e) Part of Steinert syndrome (14B.35) Isolated Haemorrhage Associated with meningococcal infection (Waterhouse-Friedrichsen syndrome)25 Other specified causes26 Idiopathic 8A.5 OMIM E27.8 A18.7, E35.1 E27.4 E27.4 E27.4 A39.1, E35.1 E27.4 E27.4 E27.4 Classified elsewhere: 6A.1 8C ADRENAL EXCESS 8C.1 Glucocorticoid excess (Cushing syndrome)27 [primary] [secondary 14C.1] Caused by ACTH excess Cushing’s disease (ACTH-producing pituitary adenoma)28 CRF excess29 Ectopic ACTH syndrome30 Increased peripheral glucocorticoid production ACTH-independent macronodular adrenal hyperplasia Multinodular adrenal hyperplasia, isolated or as part of the Carney complex31 Adrenal adenoma32 8C.1a 8C.1a.1 8C.1a.2 8C.1a.3 8C.1b 8C.1b.1 8C.1b.2 8C.1b.3 ESPE Classification of Paediatric Endocrine Diagnoses E24 E24.0 219090 E24.0 E24.3 E24.3 E24.8 #219080 E24.8 #160980 E24.8 E24.8 56 ESPE Code Diagnosis 8C.1c.z Adrenal carcinoma33 Glucocorticoids from other sources Iatrogenic Cushing syndrome34 Other 8C.2 Virilising and feminising adrenal tumours35 8C.3 8C.3b Mineralocorticoid excess Conn syndrome36 Substances with mineralocorticoid action (liquorice)37 8D DISORDERS OF THE ADRENAL MEDULLA 8D.1 Adrenal medulla defect Haemorrhage Infection Tumour Idiopathic 8C.1b.4 8C.1c 8C.1c.1 8C.3a 8D.1a 8D.1b 8D.1c 8D.1z 8D.2 8D.2a 8D.2b Primary tumour in adrenal medulla Phaeochromocytoma38 Neuroblastoma39 OMIM ICD10 E24.8 E24.8 E24.2 E24.2 E26.0 E26.0 E26.0 A39.1, E35.1 E35.1 E35.1 E35.1 M8700/0 M9500/3 1 Cholesterol side-chain cleavage deficiency (congenital lipoid adrenal hyperplasia; P450scc) Phenotype: Deficiency of all gonadal and adrenal steroid hormones including progesterone, which is required to maintain normal pregnancy. Rare cases survive untreated for several years. Life-threatening adrenal insufficiency and sex reversal in 46,XY individuals. Comments: Disruption of the P450 side-chain cleavage enzyme. Autosomal-recessive mutations of the CYP11A1 gene. Heterozygous carriers are healthy and fertile. 2 3beta-Hydroxysteroid dehydrogenase deficiency Phenotype: Various degrees of salt wasting, premature pubarche and hirsutism in girls and incomplete masculinisation in boys, growth acceleration, advanced bone age. Heterogenous clinical presentation. 17alpha-OHP (–), DHEAS (+), 17alpha-hydroxypregnenolone (+). Comments: Mutations in the type II 3beta-HSD gene. Wide spectrum of mutations associated with different phenotypic manifestations. 3 21-Hydroxylase deficiency (P450c21) (21-OHD) Comment: Most common cause of congenital adrenal hyperplasia. Adrenal Disorders 57 4 Salt-wasting 21-OHD Phenotype: Most severe form of CAH with total deficiency of enzymes of cortisol and aldosterone biosynthesis. In female newborns masculinised external genitalia (ambiguous genitalia with clitoral enlargement and urogenital sinus). Life-threatening salt wasting crises, Na (–), K (+), cortisol (–), ACTH (+), 17OHP (+), androstenedione (+). Accelerated growth and advanced bone age, peripheral precocious puberty, early epiphyseal closure and reduced final height. Comments: CYP21 gene deletion/conversion that totally ablates enzyme activity. Approximately 30% of CAH cases. Autosomal-recessive inheritance. 5 Simple virilising 21-OHD Phenotype: Milder form of CAH without salt wasting crisis, adequate aldosterone synthesis. Comment: Mutations in the CYP21 gene with 1–2% of normal enzyme activity, mainly I172N mutations. 6 Non-classical 21-OHD (late onset CAH) Phenotype: Symptoms of hirsutism and early pubertal onset, advanced bone age. Comment: Mildest form of CAH with 20 to 60% of enzyme activity, e.g. V281L and P30L mutations. In compound heterozygous forms of CAH the milder defect predicts the phenotype. 7 11beta-Hydroxylase deficiency (P450c11) Phenotype: Androgen excess and hypertension. In females ambiguous genitalia, virilisation, clitoromegaly. Accelerated growth, advanced bone age. Mineralocorticoid excess, sodium retention, volume expansion. Cortisol (–), ACTH (+), deoxycortisol (+), deoxycorticosterone (+), PRA (–), aldosterone (–), androgens (+). Comments: Second most common form of CAH. Mutations in CYP11B1 gene. Autosomal recessive. 8 Corticosterone methyl oxidase deficiency type I Phenotype: Caused by disorders of P450c11AS, the isoenzyme of P450c11, that is exclusively expressed in the zona glomerulosa. Aldosterone (–), 18-OH corticosterone (–), corticosterone (+), ACTH (+), cortisol (N). Comment: Autosomal-recessive inheritance. 9 Corticosterone methyl oxidase deficiency type II (aldosterone synthase deficiency) Phenotype: Congenital hypoaldosteronism, life-threatening salt loss, failure to thrive. Na (–), K (+), aldosterone (–), renin (+), cortisol (N). Comment: Mutations in the CYP11B2 gene encoding aldosterone synthase (P450c11Aldo), under control of the renin-angiotensin system. Autosomal-recessive inheritance. 10 Glucocorticoid suppressible hypertension (glucocorticoid remediable aldosteronism, GRA) Phenotype: Hypertension of variable degree, familial hyperaldosteronism type I. Aldosterone (+), ACTH (+). Only partial suppression of ACTH by glucocorticoids required for correction of hypertension. Comment: ACTH-regulated hybrid CYP11B1/CYP11B2 gene. ESPE Classification of Paediatric Endocrine Diagnoses 58 11 17alpha-Hydroxylase/17/20 lyase deficiency (P450c17) Phenotype: Hypertension, undervirilised genitalia in males, lack of pubertal development, salt retention. Cortisol (–), ACTH (+), deoxycorticosterone (+), T (–), estrogens (–), renin (–), K (–), alkalosis. Comment: Mutations in CYP17 gene, autosomal recessive. 12 P450 oxidoreductase deficiency (may be part of Antley Bixler syndrome) Phenotype: Skeletal defects like craniosynostosis and radiohumeral synostosis, ambiguous genitalia. In addition, cytochrome P450 enzymes affected and abnormalities in steroidogenesis (17alphahydroxylase, 17,20-lyase, 21-hydroxylase). Cortisol (–), 17OHP (+), adrenal androgens (–). Comments: Mutations of POR gene encoding NADPH: cytochrome P450 oxidoreductase (CYPOR) in patients with Antley Bixler syndrome. Mutations in FGFR2 gene in some patients described. 13 Glucocorticoid receptor defect Phenotype: Hirsutism, fatigue, menstrual disorders, obesity, hypokalaemic hypertension. Cortisol (+), adrenal androgens (+), ACTH (+). Comments: Mutations in the glucocorticoid receptor gene. 14 DAX-1 mutation (familial hypogonadotrophic hypogonadism and adrenal hypoplasia congenita) Phenotype: X-linked congenital adrenal hypoplasia and lack of pubertal development, abnormalities in spermatogenesis. Variable clinical presentation; in female carriers delayed puberty. LH (–), FSH (–), T (–), glucocorticoid and mineralocorticoid deficiency. Comments: Deletion or mutation of the NROB1 (DAX1) gene, encoding DAX1, an orphan nuclear receptor. DAX1 expression has been shown in regions of the hypothalamo/pituitary/adrenal/gonadal axis. 15 Adrenoleukodystrophy and adrenomyeloneuropathy (X-ALD) Phenotype: Inherited neurometabolic disease associated with demyelination of the central nervous system, adrenal insufficiency, and accumulation of very long chain fatty acids in plasma and tissues. Clinically heterogeneous disorder ranging from severe childhood cerebral form to asymptomatic persons. Adrenomyeloneuropathy and adrenoleukodystrophy are different presentations of the same single gene disorder. Frequently associated with adrenal insufficiency (Addison’s disease) which may remain the only clinical expression of ALD. Comment: Mutations in the ABCD1 gene encoding the adrenoleukodystrophy protein. 16 Primary xanthomatosis (Wolman’s disease) Phenotype: Rapidly fatal lysosomal storage disease caused by complete absence of lysosomal acid lipase activity. Failure to thrive, anaemia, hepatomegaly, enlarged and calcified adrenal glands and decreased cortisol secretion develop during the first months of life. Successful cure by umbilical cord stem cell transplantation has been reported. Comments: Autosomal-recessive inheritance. Gene encoding lysosomal acid lipase deficiency mapped to chromosome 10q23.2-q23.3. Adrenal Disorders 59 17 Familial glucocorticoid resistance Phenotype: Partial end-organ resistance to glucocorticoids; variable phenotype from asymptomatic to severe hyperandrogenism (sex reversal in females), fatigue and/or mineralocorticoid excess, hypertension. Cortisol (+), ACTH (+), mineralocorticoids (+), androgens (+). Comment: Mutations in the glucocorticoid receptor-alpha (hGRalpha) which impair normal glucocorticoid signal transduction, familial and sporadic cases. 18 Triple A (Allgrove) syndrome Phenotype: Rare autosomal-recessive disorder characterised by ACTH-resistant adrenal failure, achalasia, alacrimia and a progressive neurological syndrome. Genotype/phenotype analysis shows high variability in occurrence, age of onset and severity of clinical symptoms even in patients with the same mutations. Comment: Mutations in the AAA gene which encodes the ALADIN protein. 19 Pseudohypoaldosteronism type 1 Phenotype: Severe salt loss by a defect in one of the subunits of the epithelial sodium channel (autosomal recessive). An autosomal-dominant form is caused by a defect in the mineralocorticoid receptor gene NR3C2. 20Autoimmune adrenalitis (Addison’s disease) Phenotype: Weakness, fatigue, weight loss, hyperpigmentation of the skin and/or mucosae, anorexia, vomiting, hypotension, hypoglycaemia. Glucocorticoid and mineralocorticoid deficiency. ACTH (+), PRA (+). May be associated with other autoimmune diseases (Hashimoto’s thyroiditis, coeliac disease, etc., autoimmune polyglandular diesease, Schmidt syndrome). Comment: Adrenal autoantibodies may be transferred transplacentally from an affected mother to the baby. 21 Tuberculosis Phenotype: Impairment of adrenal function may occur in active tuberculosis. Also primary adrenal tuberculosis. Comment: Rifampicin treatment has an effect on adrenal steroid metabolism. 22 Fungal infections Phenotype: Histoplasmosis or coccidiomycosis may cause Addison’s disease. 23 Bacterial sepsis Phenotype: Adrenal crisis may occur during an acute meningococcal, pneumococcal, streptococcal or Haemophilus infection. Waterhouse-Friderichsen syndrome in meningococcal sepsis with meningitis: massive adrenal haemorrhage with very poor prognosis. 24 AIDS Phenotype: High incidence of adrenal insufficiency in critically ill HIV patients. ESPE Classification of Paediatric Endocrine Diagnoses 60 25 Associated with meningococcal infection (Waterhouse- Friderichsen syndrome) Phenotype: Acute adrenal insufficiency occurring with meningococcaemia. High fever, circulatory collapse, skin haemorrhage, subcapsular adrenal haemorrage. Very high mortality. 26 Haemorrhage – other specified causes Phenotype: Adrenal haemorrhage of the newborn may occur after prolonged labour and traumatic delivery. Acute shock, hypoglycaemia, hyponatraemia, hyperkalaemia, acidosis. 27 Glucocorticoid excess (Cushing syndrome) Phenotype: Truncal obesity and moon facies, acne, hypertension, striae, loss of muscle mass, decreased growth velocity, emotional instability. Cortisol (+), androgens (+), estrogens (N/+), aldosterone (+), ACTH (+/N/–). Elevated blood glucose, hyperlipidaemia, insulin-resistant diabetes. Comment: Symptoms of hypercortisolism are similar whatever its cause. 28 Cushing’s disease (ACTH-producing pituitary adenoma) Phenotype: See 8C.1. ACTH (+), cortisol (+), adrenal androgens (+), urinary 17-hydroxycorticosteroid (+), urinary free cortisol (+). ACTH and cortisol response to CRH stimulation and suppression to dexamethasone indicates pituitary-dependent disease. MRI of the brain, eventually with sinus petrosus sampling for exact localisation of the tumour. May occur as a rare manifestation of MEN1. 29 CRF excess Phenotype: See 8C.1. ACTH (+), cortisol (+). Ectopic tumors secreting CRH (e.g. phaeochromocytoma, ganglioneuroblastoma). Most CRH-producing tumours also secrete ACTH. 30 Ectopic ACTH syndrome (EAS) Phenotype: See 8C.1. Ectopic, nonpituitary ACTH secretion. ACTH (+), cortisol (+). Dynamic tests not very reliable. Bilateral inferior sinus petrosus sampling shows absent central gradient. Imaging discovers 2/3 of cases. Bronchial carcinoid, neuroendocrine tumors, gastrinomas, etc. In 20% no source of EAS detectable. 31 Multinodular adrenal hyperplasia, isolated or as part of the Carney complex. Phenotype: Primary adrenocortical hyperplasias leading to Cushing syndrome include primary pigmented nodular adrenocortical disease and ACTH-independent macronodular adrenal hyperplasia. Carney complex is a familial multiple endocrine neoplasia syndrome characterised by spotty skin pigmentation, myxomas, endocrine overactivity and schwannomas. Cortisol (+), ACTH (–), impaired response to CRH stimulation and dexamethasone suppression. Comments: Carney complex inherits autosomal dominant. Mutations in the PRKAR1A gene coding for protein kinase A. Somatic mutations in adrenal neoplasms. 32 Adrenal adenoma Phenotype: See 8C.1. Autonomous production of cortisol by an adrenal adenoma. Cortisol (+), adrenal androgens (+), ACTH (–). No response of ACTH and cortisol to CRH stimulation and dexamethasone suppression. Comment: Adrenal masses may be detected incidentally by imaging procedures (incidentaloma), approximately 10% produce glucocorticoids. Adrenal Disorders 61 33 Adrenal carcinoma Phenotype: See 8C.1. Hormonally active tumours. Cortisol (+), ACTH (–). 34 Iatrogenic Cushing syndrome Phenotype: See 8C.1. Oral, parenteral, inhaled, topical or intra-articular administration of glucocorticoids causes Cushing syndrome in a dose-dependent manner. Adrenal suppression, osteoporosis, growth retardation. Cortisol (+ or –), ACTH (–). 35 Virilising and feminising adrenal tumours Phenotype: Virilising tumours: peripheral precocious puberty, virilisation, accelerated growth. T (+), cortisol (N/+). LH, FSH (N), but no response to LHRH stimulation. Diagnostic imaging. Adenoma or carcinoma. Feminising tumours: peripheral precocious puberty, gynaecomastia in boys. E2 (+), unresponsiveness of LH and FSH to GnRH stimulation. 36 Conn syndrome (primary hyperaldosteronism) Phenotype: Hypertension, pollakisuria, muscular weakness, tetany. Na (+), K (N/–), metabolic alkalosis. Aldosterone (+), renin (–). Primary aldosteronism is caused by bilateral hyperplasia in 2/3 cases and by aldosterone-producing adenoma in 1/3. Imaging and adrenal vein sampling to distinguish unilateral and bilateral adrenal aldosterone production. Comment: Subtype of glucocorticoid-suppressible aldosteronism. Elevated aldosterone synthase which increases 18-hydroxycortisol in plasma. See 8A.1e.3. 37 Substances with mineralocorticoid action (liquorice) Phenotype: Hypertension, hypokalaemia, aldosterone (–), renin (–). Liquorice contains glycyrrhizic acid, which inhibits 11-hydroxysteroid dehydrogenase activity. 38 Phaeochromocytoma Phenotype: Hypertension, sweating, headache, palpitations, weight loss, polyuria, pallor/flushing. Epinephrine (+), norepinephrine (+) in plasma and urine. Imaging procedures for exact localisation, 90% from adrenomedullary tissue, <10% malignant. Frequently familial. 39 Neuroblastoma Phenotype: Hypertension and flushing are found rarely. Symptoms and signs depend largely on size and localisation of the tumour and whether it has metastasised (lymph nodes, liver, skin, bone, bone marrow). Excretion of catecholamines (+), dopamine (+), vanillylmandelic acid (+). Localisation by imaging procedures. High rate of spontaneous regression. Mainly in children <5 years. Comments: Several chromosomal loci show loss of heterozygosity, indicative of a tumour suppressor gene. Gain of the long arm of chromosome 17q is the most frequent chromosomal abnormality. ESPE Classification of Paediatric Endocrine Diagnoses 62 ESPE Code Diagnosis 9 OMIM ICD10 TESTI CU L AR D ISO R D E R S/ D I SO R D E R S O F M ALE G E N ITAL S Excluded: Testicular hyperfunction/testotoxicosis (3A.2c.2) Congenital defects with malformation of external genitalia resulting in sexual ambiguity (4B.1, 4B.2) 9A HYPERGONADOTROPHIC HYPOGONADISM (primary testicular failure) E29 Possible secondary codes: 3C.1 (feminisation/gynaecomastia) 3D.0 (delayed puberty) 9A.0 Due to disorder classified elsewhere Cryptorchidism (9B) Klinefelter syndrome and its variants (14A.3) Laurence-Moon-Bardet-Biedl syndrome [primary 14B.18] [secondary 5b.2a] Noonan syndrome (14B.24) Steinert myotonic dystrophy syndrome (14B.35) 9A.1 Congenital disorders of the testes not leading to a disorder of sex development Excluded: 46,XY disorders of sex development (4B) Disorders classified elsewhere: 45,X/46,XY mixed gonadal dysgenesis with normal male genitalia (14A.4) 46XY gonadal dysgenesis Isolated elevation of FSH1 (Sertoli-cell-only syndrome/FSH receptor defect, spermatogenic arrest)2 Androgen receptor defect without malformation of external genitalia3 Anorchia (vanished testes, testicular regression syndrome)4 Other specified congenital testicular disorders leading to abnormal postnatal sex development Other congenital testicular disorders, unspecified, leading to abnormal postnatal sex development 9A.1a 9A.1b 9A.1c 9A.1d 9A.1e 9A.1y 9A.1z 9A.2 E29.8 E29.8 #400042 E29.8 #300068 E34.5 273250 Q55.0 E29.8 E29.9 Acquired forms of testicular failure Testicular Disorders/Disorders of Male Genitals 63 ESPE Code Diagnosis 9A.2z Disorders classified elsewhere: Autoimmune (part of autoimmune polyglandular syndrome type 1, 14C.4a) (Post-)infection5 Spontaneous torsion Traumatic Iatrogenic6 After scrotal or inguineal surgery After irradiation After chemotherapy Other specified disorders Other disorders, unspecified 9B CRYPTORCHIDISM/MALDESCENDED TESTES7 9A.2a 9A.2b 9A.2c 9A.2d 9A.2e 9A.2e.1 9A.2e.2 9A.2e.3 9A.2y OMIM ICD10 E29.1, E89.5 E29.8 E29.8 E29.8 E29.1 E89.5 E89.5 E89.5 E29.1 E29.1 #219050 Q53 Note: If secondary to an endocrine disorder then use code 9B as supplementary code Unilateral Suprascrotal Inguinal Abdominal Ectopic Q53.1 Q53.2 9B.2e Bilateral Suprascrotal Inguinal Abdominal Ectopic Combination of various locations 9B.3 Retractile testes Q55.2 9B.4 Acquired cryptorchidism (testis has definitely been in scrotum on previous examinations, but later found to be suprascrotal or inguineal, needing treatment) Q53.9 9C ACQUIRED TESTICUL AR DISORDERS 9C.1 Orchitis If associated with testicular failure then also give code 9A.2b 9B.1 9B.1a 9B.1b 9B.1c 9B.1d 9B.2 9B.2a 9B.2b 9B.2c 9B.2d ESPE Classification of Paediatric Endocrine Diagnoses N45 64 ESPE Code Diagnosis 9C.2 Testicular torsion8 If associated with testicular failure also give code 9A.2c 9C.8 Other, specified disorders of testes (e.g. trauma, iatrogenic) If associated with testicular failure also give appropriate codes 9A.2d or 9A.2e OMIM ICD10 75 N44 S30.8 S39.9 9D TUMOURS OF TESTES Benign: D29.2 Malignant: C62 9D.1 Germ cell origin Seminomas Non-seminomas Carcinoma in situ Yolk-sac tumours (embryonal cell tumours) Teratoma Choriocarcinoma M906–M909 Non-Germ Cell Origin Leydig cell tumour Sertoli Cell tumour Primitive gonadal structures M859–M867 9D.1a 9D.1b 9D.1b.1 9D.1b.2 9D.1b.3 9D.1b.4 9D.2 9D.2a 9D.2b 9D.2c 9D.3 9D.3a 9D.3z Mixed tumours Gonadoblastomas Other mixed testicular tumours 9D.8 Other, specified tumours of testes (e.g. leukaemia, rhabdomyosarcoma) 9E DISORDERS OF PENIS M9073/1 C62.9 C62.9 Note: If due to underlying endocrinological disorder then use code 9E only as secondary code 9E.1 9E.1a 9E.1b 9E.1c Hypospadias9 Glandular hypospadias Penile hypospadias Penoscrotal hypospadias Testicular Disorders/Disorders of Male Genitals Q54 Q54.0 Q54.1 Q54.2 65 ESPE Code Diagnosis OMIM ICD10 9E.1z Other Q54.8 9E.2 Epispadias Q64.0 9E.3 Cloacal malformation with penile abnormality Q43.7 9E.4 Microphallus (micropenis)10 Q55.6 9F S C R O TA L D I S O R D E R S 9F.1 Bifid scrotum Q55.2 9F.2 Shawl scrotum (if part of Aarskog-Scott syndrome, use 14B.1 as [primary], and 9F.2 as [secondary]) N50.9 9G D I SO R D E R S O F TH E E PI D I DYM I S 9G.1 Epididymitis N45 9G.2 Spermatocele Q55.4 9G.3 Discontinuity of efferent ducts or vas deferens Q55.4 9H DISORDERS OF TESTICULAR BLOOD VESSELS 9H.1 Varicocele 9H.9 Other disorders, unspecified 9Y OTHER SPECIFIED DISORDERS OF THE MALE G E N I TA L I A Q55.8 9Z O T H E R D I S O R D E R S O F T H E M A L E G E N I TA L I A , UNSPECIFIED Q55.9 ESPE Classification of Paediatric Endocrine Diagnoses I86.2 66 1 Isolated elevation of FSH (Sertoli-cell-only syndrome (SCO)/FSH receptor defect) Synonym: Del Castillo syndrome. Phenotype: Infertility, small testes in adulthood. Histology: Small seminiferous tubules without germ cells. Adulthood: FSH (+), inhibin B (–). 2 Spermatogenic arrest Phenotype: Infertility, small to normal sized testes in adulthood. Histology: Seminiferous tubules of reduced diameter; arrest of spermatogenesis at spermatogonial, spermatocytic or spermatid level; no secondary spermatids. Adulthood: FSH (+). 3 Androgen receptor defect without malformation of external genitalia Phenotype: Normal or small penis. Infertility, small to normal sized testes in adulthood. Histology: Quantitative and qualitative defect of spermatogenesis. Adulthood: FSH (N, +, –), LH (N, +), T (N, +). 4 Anorchia, vanished testes, testicular regression syndrome Synonym: XY gonadal agenesis syndrome, testicular atrophy/aplasia. The term ‘vanishing testes’ is also used, but this should be reserved for the rare cases when (small) testis had been present at birth, but later vanished without known cause, suggesting a continuing process. Phenotype: Normal penis, empty scrotum, absence of puberty. FSH (+), LH (+), T (–). 5 (Post-)infectious secondary testicular failure Phenotype: Reduced fertility. Atrophy of seminiferous tubules; Leydig cells intact. Adulthood: FSH (+). 6 Iatrogenic, e.g. post-irradiation, post-chemotherapy testicular failure Phenotype: Defective pubertal development, infertility. Leydig cell failure may or may not occur, depending on the severity of damage. Histology: Sertoli-cell-only, atrophy of seminiferous tubules, atrophy of interstitial tissue including Leydig cells. Adulthood: FSH (+), LH (N, +), T (N,–). 7 Cryptorchidism/maldescended testes Synonym: Retentio testis, undescended testes, cryptorchism. Phenotype: Unilateral or bilateral empty scrotum. Comment: Retractile testes are classified under the general heading of cryptorchidism, although some may consider this as a variant of normal. 8 Testicular torsion Phenotype: Painful swelling of the testis. Occlusion of blood supply to the testis. Comment: Bilateral intrauterine torsion results in anorchia; postnatal torsion results in primary testicular failure. 9 Hypospadias Phenotype: Glandular, penile or penoscrotal hypospadias. Comment: Occurs in higher frequency in hypogonadotrophic hypogonadism. 10 Microphallus (micropenis) Phenotype: Small but otherwise normally shaped penis, measuring less than –2 SDS (25 mm at birth) in stretched length from the pubic bone to the tip of the glans penis. For review of anthropometric measurements, see Hughes et al. [Arch Dis Child 2006;91:554–563]. Testicular Disorders/Disorders of Male Genitals 67 ESPE Code Diagnosis OMIM ICD10 10 OVAR I ES , FE MALE R E PRODUC TIVE TR AC T AN D B R E A STS 10A OVARY E28.3 10A.1 Primary ovarian failure (hypergonadotrophic hypogonadism) Possible supplementary codes: 3E.1a.0 primary amenorrhoea 3E.1b.0 secondary amenorrhoea Due to disorder classified elsewhere: Steroidogenic block: CAH (8A.1) Aromatase deficiency (4C.2b) Turner syndrome (14A.5) Autoimmune polyglandular syndrome (14C.4a) Gonadal agenesis Gonadal dysgenesis Pure 46,XX gonadal dysgenesis (complete or incomplete)1 Gonadal dysgenesis with other specified chromosomal/genetic abnormality (e.g. trisomy 13, trisomy 18, trisomy 21, Denys Drash syndrome in XX individual) Mixed gonadal dysgenesis2 Other, specified gonadal dysgenesis, e.g. 47,XXX, etc. E28.3 10A.1a 10A.1b 10A.1c 10A.1c.1 10A.1c.2 10A.1c.3 10A.1c.8 600171 Q99.1 Q99.1 Q99.1 Q99.8 Q97.0 Q97.1 Q97.2 Q97.8 10A.1c.9 Gonadal dysgenesis, unspecified Q98.9 10A.1d E89.4 10A.1z Post-ablative ovarian failure, e.g. post-irradiation, post-surgical, post-chemotherapy3 Resistant ovary syndrome (Savage syndrome, mutation of FSH receptor gene)4 Due to other specified disorder, e.g. infection/oophoritis, autoimmune SLE5 Idiopathic/unspecified 10A.2 Ovarian androgen excess 10A.1e 10A.1y ESPE Classification of Paediatric Endocrine Diagnoses E28.3 *136435 E28.3 E28.3 E28.3 E28.1 68 ESPE Code Diagnosis 10A.2a 10A.2b 10A.3 10A.3a 10A.3b 10A.3c 10A.3d Polycystic ovary syndrome6 Other causes OMIM ICD10 #184700 E28.2 E28.2 Ovarian cysts and tumours Ovarian follicular cyst Note: if associated with precocious pseudopuberty: 3A.2c.1 Corpus luteum cyst Cysts, unspecified Germ cell tumours7 N83.0 N83.1 N83.2 Benign: D27 Malignant: C56 10A.3e Non-germ cell tumours Benign: D27 Malignant: C56 10A.3e.2 Granulosa tumour8 Other specified tumours 10B DISORDERS OF THE UTERUS AND CERVIX 10A.3e.1 Functional disorders are classified in 3E, menstrual disorders 10B.1 10B.1a 10B.1b 10B.1c 10B.1d 10B.1y Congenital malformations Agenesis and aplasia of the uterus (Müllerian agenesis/MayerRokitansky-Kuster-Hauser syndrome, Müllerian-renal-cervical spine (MURCS) syndrome)9 Congenital absence of the cervix (isolated) Endometrial hypoplasia/aplasia Incomplete Müllerian fusion [includes: double uterus (uterus didelphy), half uterus (uterus unicornis), partial duplication (uterus bicornis, Fryns syndrome), partial or complete uterine septum (uterus septus and subseptus)] Other specified congenital malformations of uterus and cervix Q51 #277000 Q51.0 Q51.5 Q51.8 192050 Q51.2– Q51.4 Q51.8 Q51.6 Q51.7 Q51.1 10B.1z Other congenital malformations of uterus and cervix, unspecified Q51.9 10B.2 Acquired disorders of the uterus and cervix Uterine synechiae/Asherman syndrome Cervical stenosis N85.6 10B.2a 10B.2b Ovaries, Female Reproductive Tract and Breasts N88.2 69 ESPE Code Diagnosis 10B.2z Other specified acquired malformations of uterus and cervix Acquired malformations of uterus and cervix, unspecified 10B.3 Tumours of uterus and cervix 10B.2y OMIM ICD10 Benign: D25, D26 Malignant: 10B.8 Other specified disorder of uterus and cervix, e.g. polyps 10B.9 Disorder of uterus and cervix, unspecified 10C D I S O R D E R S O F T H E VAG I N A AN D E X T E R N AL F E M A L E G E N I TA L I A C53, C54 N84.0 N84.1 Possible secondary codes: 3E.1a.0 primary amenorrhoea 3E.1b.0 secondary amenorrhoea 10C.1 10C.1a 10C.1b 10C.1c 10C.1d 10C.1e 10C.1y 10C.1z 10C.2 10C.2a Congenital malformations Vaginal agenesis (isolated) Imperforate hymen (can be part of McKusick-Kaufman syndrome) Transverse vaginal septum Labial fusion/agglutination Congenital malformation of the clitoris Excluded: Clitoromegaly due to endocrine causes/virilisation (4C.2) Other specified congenital malformations of the female external genitalia and vagina Congenital malformations of female external genitalia and vagina, unspecified Acquired disorders of the vagina and external female genitalia Adhaesions (vaginal, labial, vulval) Q52 Q52.0 #236700 Q52.3 Q52.8 Q52.5 Q52.6 Q52.8 Q52.9 N89.5 N90.8 10C.2b 10C.2c 10C.2z Acquired disorders of the clitoris Excluded: Clitoromegaly due to endocrine causes (3C.2) Trauma Other disorders, unspecified ESPE Classification of Paediatric Endocrine Diagnoses N90.8 S30.2 S30.2 70 ESPE Code Diagnosis 10C.3 OMIM Tumours of the vagina and external female genitalia ICD10 Benign: D28.0 D28.1 D28.7 D28.9 Malignant: C51, C52 10C.8 Other, specified, disorders of the vagina and external female genitalia E.g. polyps 10C.9 Disorders of the vagina and external female genitalia, unspecified 10D DISORDERS OF THE BREAST 10D.1 Galactorrhoea (not associated with childbirth) Excluded: Galactorrhoea in the male (3C.1) 10D.2 Disorders of size Hypoplasia/aplasia/hypomastia/micromastia Macromastia 10D.2a 10D.2b 10D.3b Disorders in numbers Polythelia, polymastia Absence of breast and nipple (athelia) 10D.4 Tumours of breasts 10D.3 10D.3a N84.2– N84.9 N64.3 Q83.8 N62 %163700 Q83.1 113700 Q83.0 Benign: D24 Malignant: C50 10D.8 Other specified disorders of the breast N60, N61 N63– N64.8 Q83.2 Q83.3 Q83.8 10D.9 Other disorder of the breast, unspecified N64.9 Q83.9 Ovaries, Female Reproductive Tract and Breasts 71 1 Pure XX gonadal dysgenesis Phenotype: Presenting as primary amenorrhoea, no dysmorphic features. FSH (+), LH (+), E2 (–). Normal female external genitalia, insufficient breast development, streak gonads. Karyotype 46,XX. 2 Mixed gonadal dysgenesis Phenotype: Somatic features of Turner syndrome, short stature; genitalia female, ambiguous or male. Gonads may be asymmetrical with streak ovaries or dysgenetic testes, also external genital development may be asymmetrical. Dysgenetic testes bear the risk of gonadoblastoma. LH (+), FSH (+), E/T (–). Comment: Karyotype commonly 45,X/46,XY. 3 Post-ablative ovarian failure, post-irradiation, post-surgery, post-chemotherapy Phenotype: Prepubertal or pubertal gonadal injury may result in hypergonadotrophic hypogonadism. Ovarian surgery (cysts, malignancy). Chemotherapy (nitrogen mustard compounds), especially sensitive during pubertal period. Pelvic irradiation (leukaemia). LH (+), FSH (+), E (–). 4 Resistant Ovary Syndrome (Mutation of FSH receptor) Phenotype: Premature ovarian failure occurs in 0.1% before the age of 30 years. Mutations have been identified in genes like inhibin alpha, FSH receptor and LH/choriogonadotrophin receptor. In addition, various transcription factors that regulate oocyte gene expression may be affected. Amenorrhoea, LH (+), FSH (+), E (–). 5 Due to other specified disorders, e.g. infection/oophoritis, autoimmune Phenotype: Disturbance of pubertal development depending on the time of onset. Mumps can cause oophoritis. Autoimmune oophoritis, usually at puberty, may be associated with autoimmune polyglandular syndrome. Autoimmune steroidal cell autoantibodies may be detected. Galactosaemia may be associated with ovarian failure (increased concentration of galactose-1-phosphate). 6 Polycystic ovary syndrome (PCO) Synonym: Stein-Loewenthal syndrome. Phenotype: Menstrual irregularity, amenorrhoea, anovulatory cycles, hirsutism, obesity, decreased fertility. Enlarged ovaries with multiple cysts, hyperthecosis. LH (+), FSH (N), LH:FSH ratio >3, T (+), adrenal androgens (+), Prl (+), insulin resistance. Comments: Not observed in prepubertal girls. No clear-cut diagnostic criteria during puberty. 7 Germ cell tumours Phenotype: Very malignant endodermal (yolk sac) tumours, occur in the upper vagina and cervix, cause bleeding. 8 Granulosa cell tumour Phenotype: Rapidly progressing virilisation, dysmenorrhoea. ⌬4 androgens (++), T (+), DHEA (N to +). 9 Agenesis and aplasia of the uterus (Müllerian agenesis/Mayer-Rokitansky-Küster-Hauser syndrome, Müllerian-renal-cervical spine syndrome (MURCS)) Phenotype: Hypoplasia of the external genitalia, agenesis of Müllerian structures, absence of vagina and hypoplasia of the uterus. Normal ovarian function. In MURCS association with renal and spinal abnormalities. Normal female karyotype. ESPE Classification of Paediatric Endocrine Diagnoses 72 ESPE Code Diagnosis OMIM ICD10 11 D ISO R D E R S O F G LUCOSE AN D TR I G LYCE R I D E M E TABO LISM 11A DIABETES ME LLITUS1 11A.1 +222100 E10 11A.1a 11A.1b Type 1 diabetes [I]2 Immune mediated [I.A] Idiopathic [I.B] 11A.2 Type 2 diabetes [II]3 #125853 E11 11A.3 Other specific types [III] Genetic defects of -cell function [III.A]4 HNF-1␣ defect (MODY3) [III.A1] Glucokinase defect (MODY2) [III.A2] HNF-4␣ (MODY1) [III.A3] Insulin promoter factor-1 (MODY 4) [III.A4] Hepatocyte nuclear factor-1beta (MODY 5) [III.A5] NeuroD1 (MODY 6) [III.A6] Mitochondrial diabetes [III.A7]5 Other (specified), e.g. Kir6.2 activating mutations [III.A8] Genetic defects in insulin action [III.B] Type IA insulin resistance [III.B1]6 Leprechaunism [III.B2] Rabson-Mendenhall syndrome [III.B3] Lipoatrophic diabetes [III.B4] Other (specified) [III.B5], e.g. Seip-Berardinelli syndrome7 Familial partial lipodystrophy (Dunnigan syndrome) Type B insulin resistance syndrome Diseases of the exocrine pancreas [III.C] Pancreatitis [III.C1] Trauma/pancreatectomy [III.C2] Neoplasia [III.C3] Cystic fibrosis [III.C4]8 Haemochromatosis (haemosiderosis) [III.C5] Fibrocalculous pancreatopathy [III.C6] Other specified disorders [III.C7] 11A.3a 11A.3a.1 11A.3a.2 11A.3a.3 11A.3a.4 11A.3a.5 11A.3a.6 11A.3a.7 11A.3a.8 11A.3b 11A.3b.1 11A.3b.2 11A.3b.3 11A.3b.4 11A.3b.5 11A.3c 11A.3c.1 11A.3c.2 11A.3c.3 11A.3c.4 11A.3c.5 11A.3c.6 11A.3c.7 Disorders of Glucose and Triglyceride Metabolism #606391 #600496 #125851 #125850 #606392 *142410 #600394 #500002 *147670 #246200 #262190 #269700 #269700 #151660 *147670 #219700 +235200 #608189 73 ESPE Code Diagnosis 11A.3c.9 Other disorders, unspecified [III.C7] Endocrinopathies [III.D] [secondary codes]. Primary codes are indicated in brackets Acromegaly [III.D1] (2B.1) Cushing syndrome [III.D2] (8C.1) Glucagonoma [III.D3] (can be part of MEN1) (14C.5a) Phaeochromocytoma [III.D4] (8D.2a) Hyperthyroidism [III.D5] (7B) Somatostatinoma [III.D6] (can be part of MEN1) (14C.5a) Aldosteronoma [III.D7] (8C.3) Other specified disorders [III.D8] Other disorders, unspecified [III.D8] Drug- or chemical-induced [III.E] Vacor [III.E1] Pentamidine [III.E2] Nicotinic acid [III.E3] Glucocorticoids [III.E4]9 Thyroid hormone [III.E5] Diazoxide [III.E6] -Adrenergic agonists [III.E7] Thiazides [III.E8] Dilantin [III.E9] ␣-Interferon [III.E10] Other [III.E11] Infections [III.F] Congenital rubella [III.F1] Cytomegalovirus [III.F2] Other [III.F3] Uncommon forms of immune-mediated diabetes [III.G] ‘Stiff-person’ syndrome (‘stiff man’ syndrome) [III.G1] Anti-insulin receptor antibodies [III.G2]10 Other [III.G3] Other genetic syndromes sometimes associated with diabetes [III.H] [secondary]. Primary codes are indicated in brackets Down syndrome [III.H1] (14A.2) Klinefelter syndrome [III.H2] (14A.3) Turner syndrome [III.H3] (14A.5) Wolfram syndrome [III.H4] (DIDMOAD) (14B.38) 11A.3d 11A.3d.1 11A.3d.2 11A.3d.3 11A.3d.4 11A.3d.5 11A.3d.6 11A.3d.7 11A.3d.8 11A.3d.9 11A.3e 11A.3e.1 11A.3e.2 11A.3e.3 11A.3e.4 11A.3e.5 11A.3e.6 11A.3e.7 11A.3e.8 11A.3e.9 11A.3e.10 11A.3e.11 11A.3f 11A.3f.1 11A.3f.2 11A.3f.3 11A.3g 11A.3g.1 11A.3g.2 11A.3g.3 11A.3h 11A.3h.1 11A.3h.2 11A.3h.3 11A.3h.4 OMIM ICD10 #102200 +131100 #171300 #275000 +131100 184850 #190685 #222300 #598500 11A.3h.5 11A.3h.6 Friedreich ataxia [III.H5] Huntington chorea [III.H6] ESPE Classification of Paediatric Endocrine Diagnoses #229300 +143100 74 ESPE Code Diagnosis OMIM 11A.3h.7 Laurence-Moon-Bardet-Biedl syndrome [III.H7] (14B.18) 245800 11A.3h.8 11A.3h.11 Myotonic dystrophy, Steinert syndrome [III.H8] (14B.35) Porphyria [III.H9] Prader-Willi(-Labhart) syndrome [III.H10] (14B.25) Other [III.H11], e.g. Alström syndrome (14B.3) 11A.4 Gestational diabetes mellitus (GDM) [IV] 11A.5 Malnutrition related or tropical diabetes [V] 11A.6 Pathological glucose tolerance11 11A.7 Diabetic complications 11A.7a 11A.7d Diabetic ketoacidosis Diabetic nephropathy Diabetic retinopathy Diabetic neuropathy 11B H Y P O G LY C A E M I A 11B.1 Hyperinsulinism Excluded: Hyperinsulinism or impaired glucose tolerance secondary to other conditions, e.g. simple obesity (5A), PCOS (10A.2a) Transient hyperinsulinism Infant of diabetic mother12 Perinatal asphyxia Rhesus disease Intrauterine growth retardation Beckwith-Wiedemann syndrome [primary 14B.5] Idiopathic Congenital (permanent) hyperinsulinism13 SUR1 mutations KIR6.2 mutations Glucokinase mutations glutamate dehydrogenase mutations Defects in the metabolism of fatty acids (SCHAD) (3alpha-hydroxyacyl-CoA dehydrogenase) Carbohydrate-deficient glycoprotein syndrome (CDG) ICD10 #209900 11A.3h.9 11A.3h.10 11A.7b 11A.7c 11B.1a 11B.1a.1 11B.1a.2 11B.1a.3 11B.1a.4 11B.1a.5 11B.1a.9 11B.1b 11B.1b.1 11B.1b.2 11B.1b.3 11B.1b.4 11B.1b.5 11B.1b.6 Disorders of Glucose and Triglyceride Metabolism #160900 #176000 #176270 #201800 E10.1 E10.2 E10.3 E10.4 P70.4 P70.1 P70.4 P70.4 P70.4 #130650 Q87.3 P70.9 E16.1 #256450 #256450 #256450 *138130 *601609 #212065 75 ESPE Code Diagnosis 11B.1b.7 Insulinomas14 Other specified causes Other causes, unspecified 11B.1b.8 11B.1b.9 11B.2 11B.2a 11B.2b 11B.3 11B.3a 11B.3b 11B.3c 11B.3d 11B.3e 11B.3f 11B.3g 11B.3z 11B.4 11B.4a 11B.4b OMIM M8151/0 E16.1 E16.1 Hormonal deficiency Disorders classified elsewhere: Growth hormone deficiency (1B.3) Cortisol deficiency: primary (adrenal defect) (8A) Secondary (ACTH deficiency) (8B and 6A.1) Noradrenaline (epinephrine) deficiency (8D.1) Glucagon deficiency15 Defects in hepatic glycogen release/storage (glycogen storage diseases) Glucose-6-phosphatase deficiency16 Glucose-6-phosphatase translocase deficiency17 Amylo 1-6 glucosidase deficiency18 Branching enzyme deficiency19 Liver phosphorylase deficiency20 Phosphorylase kinase deficiency21 Hepatic glycogen synthase deficiency22 Other defects, unspecified Defects in gluconeogenesis Hereditary fructose intolerance, fructose-1,6-bisphosphatase deficiency23 Phosphoenolpyruvate carboxykinase (PEPCK) deficiency24 ICD10 E16.1 E74.0 +232200 +232400 +232400 #232500 +306000 +306000 #240600 E74.1 +229600 +261680 +261650 11B.4c 11B.4z 11B.5 11B.5a 11B.5b 11B.5c 11B.5d 11B.5e 11B.5f 11B.5g Pyruvate carboxylase deficiency25 Other defects, unspecified Defects of fatty acid oxidation and carnitine metabolism Very-long-chain acyl-CoA dehydrogenase (VLCAD) deficiency26 Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency27 Short-chain acyl-CoA dehydrogenase (SCAD) deficiency Long-/short-chain-L-3-hydroxy-acyl CoA (L/SCAD, LCHAD) deficiency (trifunctional protein deficiency)28 Carnitine deficiency29 Carnitine palmitoyltransferase type I deficiency30 Carnitine palmitoyltransferase type II deficiency31 #266150 E16.2 #201475 #201450 #201470 #609015 #212140 #255120 #255110 #608836 #600649 ESPE Classification of Paediatric Endocrine Diagnoses 76 ESPE Code Diagnosis OMIM 11B.5h Carnitine-acylcarnitine translocase deficiency32 Other defects, unspecified +212138 11B.5z 11B.6 11B.6a 11B.6b 11B.6c 11B.6z 11B.7 11B.7a Defects in ketone body synthesis/utilisation HMG CoA-synthase deficiency/HMG CoA-lyase deficiency Succinyl-CoA: 3-oxoacid CoA-transferase (SCOT) deficiency Hepatic glycogen synthase deficiency Other defects, unspecified Other metabolic conditions Organic acidaemias (propionic/methylmalonic) ICD10 E16.2 #605911 #245050 *138571 E71.1 #606054 #251000 11B.7b 11B.7c 11B.7d 11B.7e 11B.7f Galactosaemia33 Maple syrup disease34 3-Hydroxy 3-methylglutaryl-CoA lyase deficiency35 3-Methylcrotonyl-CoA carboxylase deficiency36 Tyrosinaemia #230400 #248600 +246450 #210200 +276700 +276600 #276710 11B.7g 11B.7h 11B.7z 11B.8 11B.8a 11B.8b 11B.8z 11B.9 11B.9a 11B.9b 11B.9c 11B.9d 11B.9e 11B.9f 11B.9y 11B.9z 11C Glutaric aciduria type 237 Mitochondrial respiratory chain complex deficiencies Other defects, unspecified #231680 #256000 Drug induced E.g. sulphonylureas, insulin, beta-blockers, salicylates, alcohol Ethanol toxicity38 Salicylate toxicity39 Toxicity from other sources, unspecified E16.0 Miscellaneous causes Idiopathic ketotic hypoglycaemia (diagnosis of exclusion)40 Infections (e.g. septicaemia, malaria) Congenital heart disease Fulminant hepatic failure 41 Reye syndrome42 Jamaican vomiting sickness 43 Other specified disorders Other disorders, unspecified E16.1 PRIMARY DISTURBANCES OF LIPOPROTEIN M E T A B O L I S M 44 Disorders of Glucose and Triglyceride Metabolism 77 ESPE Code Diagnosis 11C.1 Genetic hyperlipoproteinaemias Familial hypercholesterolaemia (type IIa hyperlipoproteinaemia)45 Heterozygous familial hypercholesterolaemia Homyzygous familial hypercholesterolaemia Familial defective apolipoprotein B-10046 Polygenic hypercholesterolaemia47 Familial combined hypercholesterolaemia (hyperlipidaemia) (type IIa)48 Familial hypertriglyceridaemia49 11C.1a 11C.1a.1 11C.1a.2 11C.1b 11C.1c 11C.1d 11C.1e 11C.2 11C.2a 11C.2b 11C.2b.1 11C.2b.2 11C.2b.3 11C.2c 11C.2d 11C.2d.1 11C.2d.2 11C.2d.3 11C.2e 11C.3 11C.3a 11C.3b 11C.3c 11C.3d 11C.3e 11C.4 11C.4a 11C.4b 11C.4c 11C.4d 11C.4e 11C.4y 11C.4z Disorders of apolipoproteins Apolipoprotein A disorders: familial APOA-I deficiency and structural APOA-I mutations50 Apolipoprotein B disorders Abetalipoproteinaemia51 Familial hypobetalipoproteinaemia52 Chylomicron retention disease53 Apolipoprotein C disorders (apolipoprotein C-II deficiency) (type Ib)54 Apolipoprotein E disorders Familial dysbetalipoproteinaemia (type III hyperlipoproteinaemia)55 Type V hyperlipoproteinaemia (HLP)56 Other apolipoprotein E disorders Lp(a) disorders57 Other disorders of lipoprotein metabolism Tangier disease58 Lipoprotein lipase deficiency (type Ia)59 Hepatic lipase deficiency Familial lecithin-cholesterol acyltransferase deficiency60 Cholesterol ester transfer protein deficiency Other disorders of lipid and cholesterol metabolism Disorders classified elsewhere: Smith-Lemli-Opitz syndrome (14B.33) Cerebrotendinous xanthomatosis61 Phytosterolaemia (beta-sitosterolaemia)62 Cholesterol ester storage disease and Wolman disease Niemann-Pick C disease Other specified disorders Other disorders, unspecified ESPE Classification of Paediatric Endocrine Diagnoses OMIM ICD10 #143890 E78.0 #144010 E78.8 E78.2 #144250 E78.0 #145750 E78.1 *107680 E78.8 +107730 E78.8 #200100 E78.6 %605019 E78.6 #246700 E78.3 #207750 E78.3 +107741 E78.1 +107741 E78.2 #144650 E78.3 +107741 E78.8 +152200 E78.8 #205400 E78.6 246650 E78.3 246650 E78.3 #245900 E78.6 #607322 E78.8 #213700 E75.5 #210250 E78.8 +278000 E75.5 #257220 E75.2 E78.8 E78.9 78 1 Diabetes mellitus Comments: For this classification the most recent updated Etiologic classification of diabetes mellitus is used [American Diabetes Association: Diagnosis and classification of diabetes mellitus. Diabetes Care 2007;30(suppl 1);S42–47]. 2 Type 1 diabetes Synonyms: Insulin-dependent diabetes mellitus (IDDM), juvenile insulin-dependent diabetes. Phenotype: Polyuria, polydipsia, nocturia, polyphagia, weight loss, or symptoms of frank diabetic ketoacidosis: air hunger, Kussmaul breathing, acetone on the breath, vomiting, dehydration, abdominal pain, obtundation of consciousness or coma. Loss of insulin secretion, acquired deficiencies of the secretion of some counterregulatory hormones (glucagon, adrenalin). Auto-antibodies: islet cells (ICA), insulin (IAA), glutamate-decarboxylase (GAD II), etc. Comment: Caused by beta-cell destruction, usually leading to absolute insulin deficiency. 3 Type 2 diabetes Synonyms: Type II diabetes, non-insulin-dependent diabetes mellitus (NIDDM). Phenotype: Non-characteristic. May be suspected in obese patients, family history positive, glycosuria, no ketonuria. Many affected children are asymptomatic, some have signs of hyperglycaemia (polyuria, polydipsia) without ketosis; the majority are overweight. Insulin ‘insufficiency’ and insulin ‘resistance’ (may range from predominantly insulin resistance with relative insulin deficiency to predominantly secretory defect with insulin resistance). Auto-antibodies (ICA, IAA, GAD II, etc.) usually negative. Comments: Probably multifactorial, familial aggregation is common. Uncommon in children, most patients are adolescents. Strongly associated with obesity of the population. 4 Genetic defects of -cell function Synonyms: Maturity-onset diabetes of the young (MODY), non-insulin-dependent diabetes of youth (NIDDY). Phenotype: Non-characteristic, strong family history with ‘mild’ diabetes at a young age; usually glycosuria, no ketonuria, no insulin resistance. Many affected children are asymptomatic or have mild symptoms, they are usually adolescents of normal weight or slightly obese. Transcription factor MODY – caused by mutations in the genes encoding hepatocyte nuclear factor (HNF)-1a (chromosome 12q, MODY 3), HNF-1b (MODY 5), HNF-4a (chromosome 20q, MODY 1) and insulin promoter factor (IPF)-1 (MODY 4) – usually results in a progressive beta-cell defect and progressive hyperglycaemia (5–25 mmol/l) with a very high and early incidence of diabetic complications. In GCK diabetes there is only mild (5.5–14 mmol/l) hyperglycaemia. Comment: Questionable in patients without NIDDM family history. 5 Mitochondrial diabetes Synonym: Maternally transmitted diabetes with deafness. Phenotype: Similar to type I or type II diabetes, deafness. Impaired insulin secretion. Comment: Maternally transmitted trait. Disorders of Glucose and Triglyceride Metabolism 79 6 Type A insulin resistance Phenotype: Type A syndrome: Extreme insulin resistance, acanthosis nigricans, hyperandrogenism without obesity or lipoatrophy. Leprechaunism: In addition to type A symptoms intrauterine and neonatal growth retardation with fasting hypoglycaemia. Lipatrophic diabetes (generalised lipodystrophy): Overall absence of fat, insulin resistance, acanthosis nigricans with or without hyperlipidaemia and sexual precocity. Typically diagnosed in young thin females before the fourth decade and sometimes by adolescence; accelerated childhood growth; acral hypertrophy; recurrent muscle cramps; skin: papillomatosis, epidermal hyperkeratosis and hyperpigmentation commonly of neck, axillae, antecubital fossae and over knuckles; virilisation with hirsutism (especially at/after puberty); obesity; menstrual abnormalities or primary or secondary amenorrhoea. Severe insulin resistance with marked glucose intolerance leading to type 2 diabetes mellitus; SHBG (–), T (+), adrenal androgens (+); hyperlipidaemia which may be life-threatening. 7 Berardinelli syndrome Synonyms: Congenital generalised lipodystrophy (CGL), total lipodystrophy, Lawrence-Seip syndrome, Berardinelli-Seip syndrome. Phenotype: Lack of adipose tissue, hypertrophy of muscle. Tall stature, large hands and feet. Phallic/ clitoral hypertrophy. Elevated basal metabolic rate. Hypertrophic cardiomyopathy. Hepatomegaly leading to cirrhosis. Acanthosis nigricans, insulin resistance, polycystic ovaries. Corneal opacities. Hyperinsulinism with insulin resistance and hyperlipidaemia. 8 Cystic fibrosis Phenotype: Typical symptoms of diabetes, ketoacidosis is rare. Chronic pancreatic inflammation. Insulin insufficiency or deficiency. 9 Secondary diabetes mellitus due to glucocorticoid therapy Synonym: Steroid diabetes. Phenotype: Symptoms of diabetes. Insulin resistance and hypoinsulinaemia (in some cases). 10 Anti-insulin receptor antibodies Synonym: Type B syndrome. Phenotype: Severe insulin resistance, acanthosis nigricans, frequently other autoimmune diseases: alopecia, vitiligo, Raynaud disease, lupus erythematosus, Sjögren syndrome, ataxia telangiectasia, arthritis, antinuclear (anti-DNA) antibodies, elevated erythrocyte sedimentation rate. Severe hyperinsulinism (10- to 100-fold of normal). 11 Pathological glucose tolerance Synonym: Impaired glucose tolerance (IGT). Phenotype: Patients are clinically asymptomatic. Insulin resistance and probably hypoinsulinaemia. Comments: Rare in children, not uncommon in obese adolescents. Persons with IGT appear to be at higher risk than the general population for the development of type 2 diabetes. 12 Infant of diabetic mother Phenotype: Large for date infant with hypoglycaemic tendency due to hyperinsulinism. Respiratory distress syndrome, polycythaemia, jaundice, hypocalcaemia and hypomagnesaemia occur more ESPE Classification of Paediatric Endocrine Diagnoses 80 commonly than in normal infants. Increased incidence of prematurity, and congenital malformations (5–8%), specifically sacral agenesis. Comment: Features of IDM can be minimised if good metabolic control of diabetes mellitus can be achieved during pregnancy. 13 Congenital hyperinsulinism Synonyms: Nesidioblastosis, hyperinsulinaemia of infancy, neonatal hyperinsulinism. Phenotype: Persistent hypoglycaemia usually beginning in the neonatal period, associated with elevated/inappropriate insulin levels. Comments: Medical therapy involves oral diazoxide and chlorothiazide (acting on KATP channel), nifedipine (acting on calcium channel). Subcutaneous somatostatin and glucagon infusions are helpful in acute management and can be given long term. 95% pancreatectomy is required for diffuse hyperinsulinism resistant to medical treatment, partial pancreatectomy for focal hyperinsulinism. Surgery should only be performed in designated specialist centres. 14 Insulinomas Phenotype: Fasting hypoglycaemia, obesity. 15 Glucagon deficiency Phenotype: Symptoms of recurrent neonatal hypoglycaemia. 16 Glucose-6-phosphatase deficiency Synonyms: Glycogenosis type Ia, von Gierke’s disease, glycogen storage disease type 1a (GSD Ia). Phenotype: Hepatomegaly, hypoglycaemia and lactic acidaemia; long-term: growth failure, osteoporosis, anaemia, gout, hyperlipidaemia, insulin resistance, hepatic adenomas, renal disease, pulmonary hypertension. 17 Glucose-6-phosphatase translocase Synonym: Glycogenosis type Ib (GSD Ib). Phenotype: As for GSD type Ia, recurrent infections, neutropenia, impaired neutrophil function, inflammatory bowel disease. 18 Amylo 1-6 glucosidase deficiency (glycogenosis type IIIa and type IIIb) Synonyms: Type IIIa: liver and muscle debrancher deficiency, limit dextrinosis, Cori or Forbes’ disease. Type IIIb: liver debrancher deficiency (GSD III). Phenotype: Type IIIa: hepatomegaly, hypoglycaemia, growth failure, muscle weakness, cardiomyopathy, hyperlipidaemia. Type IIIb: no muscle symptoms. 19 Branching enzyme Synonyms: Glycogen storage disease type IV, branching enzyme deficiency, amylopectinosis, Andersen’s disease (GSD IV). Phenotype: Failure to thrive, hepatosplenomegaly, portal hypertension, muscle weakness, cardiomyopathy, progressive stenosis, elevated transaminase levels. Disorders of Glucose and Triglyceride Metabolism 81 20 Liver phosphorylase deficiency Synonyms: Glycogen storage disease type VI, Hers disease (GSD VI). Phenotype: Hepatomegaly, diminishing with age, growth retardation, mild hypoglycaemia and hyperlipidaemia, ketosis. 21 Phosphorylase kinase deficiency Synonym: Glycogen storage disease type IX (GSD IX). Phenotype: Hepatomegaly, growth retardation, mild muscle weakness, mild hypoglycaemia, mild hyperlipidaemia. A few cases have been reported with isolated involvement of muscle or heart. Comment: Sometimes included in GSD type VI. 22 Hepatic glycogen synthase deficiency Synonym: Glycogen synthetase deficiency. Phenotype: Early morning drowsiness, fatigue, fasting hypoglycaemia, ketosis, postprandial hyperglycaemia. 23 Hereditary fructose intolerance, fructose-1,6-bisphosphatase deficiency Synonym: Fructosaemia, fructose-1,6-diphosphatase deficiency. Phenotype: Symptoms after exposure to fructose. Neonate: Poor feeding, vomiting, hypoglycaemia, hepatomegaly, liver failure, bleeding, renal tubular dysfunction. Older child: Episodic, provoked by fasting: hepatomegaly, poor growth, aversion for sweet food. Hyperventilation due to acidosis, hypoglycaemia, encephalopathy. 24 Phosphoenolpyruvate carboxykinase (PEPCK) deficiency Phenotype: Lactic acidaemia, hypoglycaemia, hypotonia, failure to thrive. Comment: Some published cases have actually had respiratory chain defects or hyperinsulinism. 25 Pyruvate carboxylase deficiency Phenotype: Type A: lactic acidaemia, hepatomegaly, mental retardation; type B: lactic acidaemia, hepatomegaly, hyperammonaemia, citrullinaemia, hyperlysinaemia, death by 3 months; type C: intermittent lactic acidaemia. Comment: Hypoglycaemia is not a prominent feature. 26 Very-long-chain acyl-CoA dehydrogenase (VLCAD) deficiency Phenotype: Fasting hypoglycaemia, cardiomyopathy, episodic rhabdomyolysis. Comment: Many cases were previously labelled as long-chain acyl-CoA dehydrogenase (LCAD) deficiency. 27 Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency Phenotype: Fasting hypoglycaemia and encephalopathy, unexpected infant death. 28 Long-/short-chain-L-3-hydroxy-acyl CoA (LCHAD) deficiency Synonym: Trifunctional protein deficiency. Phenotype: Fasting hypoglycaemia and encephalopathy, hypertrophic cardiomyopathy, myopathy and episodic rhabdomyolysis, pigmentary retinopathy and neuropathy (long-term), acute fatty liver of pregnancy or HELLP syndrome (women carrying an affected foetus). ESPE Classification of Paediatric Endocrine Diagnoses 82 29 Carnitine deficiency Synonym: Carnitine transport defect. Phenotype: Cardiomyopathy, muscle weakness, fasting hypoglycaemia. Comment: Secondary carnitine deficiency occurs in many -oxidation defects. 30 Carnitine palmitoyltransferase type I (CPT I) deficiency Phenotype: Fasting hypoglycaemia and encephalopathy. 31 Carnitine palmitoyltransferase type II (CPT II) deficiency Phenotype: Infants: fasting hypoglycaemia and encephalopathy, cardiomyopathy, congenital anomalies (kidneys, brain); adolescents, young adults: myopathy and episodic rhabdomyolysis. 32 Carnitine-acylcarnitine translocase deficiency Phenotype: Hypoketotic hypoglycaemia and encephalopathy, hyperammonaemia, cardiomyopathy, heart block, arrhythmias. 33 Galactosaemia Phenotype: Diarrhoea, vomiting, failure to thrive, cataracts, liver disease, intellectual retardation (especially affecting speech). Ovarian failure. Comment: Probably only causes hypoglycaemia as a complication of severe liver failure. 34 Maple syrup disease (MSUD) Phenotype: Neonatal: lethargy, anorexia, encephalopathy leading to seizures and coma, ketosis, odour; intermittent: episodic encephalopathy; intermediate: developmental delay and/or fits, episodic ketoacidosis. 35 3-Hydroxy 3-methylglutaryl-CoA (HMG-CoA) lyase deficiency Phenotype: Recurrent episodes of encephalopathy, acidosis and hypoketotic hypoglycaemia, liver dysfunction. 36 3-Methylcrotonyl-CoA carboxylase deficiency Synonym: 3-Methylcrotonylglycinuria. Phenotype: Recurrent episodes of encephalopathy, acidosis and hypoketotic hypoglycaemia, liver dysfunction. 37 Glutaric aciduria type 2 Synonyms: Glutaric acidaemia type II, multiple acyl-CoA dehydrogenase deficiency (severe or mild), ethylmalonic-adipic aciduria (late onset cases). Phenotype: Neonatal onset with congenital anomalies: hypotonia, metabolic acidosis, odour; hypotonia; hepatomegaly; renal cysts, dysmorphism. Neonatal onset without congenital anomalies: hypoglycaemia, metabolic acidosis, odour; hepatomegaly; cardiomyopathy. Late onset cases: Episodic vomiting, hypoglycaemia and acidosis; myopathy. 38 Ethanol toxicity Phenotype: In children, alcohol can cause hypoglycaemia after relatively short fasts. Other symptoms of ethanol ingestion: confusion, vomiting, inebriation. Disorders of Glucose and Triglyceride Metabolism 83 39 Salicylate toxicity Synonym: Aspirin toxicity. Phenotype: Hypoglycaemia can occur with overdoses or with therapeutic doses in young children (<3 years), especially if fasting. Other symptoms of toxicity: tinnitus, vomiting, tremor, sweating, confusion, hyperventilation. 40 Idiopathic ketotic hypoglycaemia (diagnosis of exclusion) Synonym: Accelerated starvation. Phenotype: Fasting hypoglycaemia, sometimes leading to seizures. Comments: This is a diagnosis of exclusion. It comprises a heterogeneous group of patients. Some may represent the lower end of the normal distribution for fasting blood glucose concentrations, whilst others may have as yet unrecognised metabolic or endocrine defects. 41 Fulminant hepatic failure Phenotype: Hypoglycaemia can occur in acute hepatic failure from any cause but only in severe cases, accompanied by encephalopathy. The clinical picture will be that of hepatic failure (jaundice, anorexia, vomiting, bleeding and encephalopathy). 42 Reye’s syndrome Phenotype: Vomiting; encephalopathy, seizures; hepatic dysfunction with hyperammonaemia and coagulopathy; hypoglycaemia; preceding viral illness and/or aspirin exposure. Comment: This is a diagnosis of exclusion, many suspected cases actually having inborn errors of metabolism (-oxidation or urea cycle defects or organic acidaemias). 43 Jamaican vomiting sickness Synonym: Hypoglycin toxicity. Phenotype: Vomiting and encephalopathy; hypoglycaemia and acidosis; liver dysfunction. Comment: Hepatic steatosis and glycogen depletion due to unripe ackee fruit (West Indies). 44 Genetic hyperlipoproteinaemias (hyperlipidaemias) Comments: In each textbook, a somewhat different classification is used. The hyperlipidaemias have traditionally been classified as Fredrickson types I–V on the basis of the type of lipoprotein particle that is increased in plasma. However, as a classification scheme it is now generally felt unhelpful. Hyperlipidaemias can be subdivided into primary (resulting from inherited defects in lipid metabolism) and secondary hyperlipidaemias (resulting from acquired metabolic conditions). In the ESPE classification the genetic hyperlipoproteinaemias are classified according to Oski’s Pediatrics, ed 4; in McMillan JA, Feigin RD, DeAngelis C, Jones MD (eds). Philadelphia, Lippincott Williams & Wilkins, 2006. 45 Familial hypercholesterolaemia (FH) Synonym: Autosomal-dominant hypercholesterolaemia, type IIa hyperlipoproteinaemia, LDL receptor disorder. Phenotype: Heterozygotes: Tendon xanthomas, xanthelasma and arcus corneae appear at the age of 20 years or later; coronary atherosclerosis from 30 years of age. Homozygotes: Cutaneous xanthomas by 4 years of age; coronary heart disease in childhood; aortic valve stenosis. ESPE Classification of Paediatric Endocrine Diagnoses 84 46 Familial defective apolipoprotein B-100 Synonyms: Autosomal-dominant hypercholesterolaemia, type B; familial defective apolipoprotein B. Phenotype: Increased risk of coronary heart disease; raised plasma cholesterol and LDL. 47 Polygenic hypercholesterolaemia Phenotype: Cholesterol and LDL are moderately elevated, lipoprotein electrophoretic pattern usually IIa type, or IIb. Comments: Multifactorial etiology. Occurs in 5% of the adult population. 48 Familial combined hyperlipidaemia Phenotype: Early coronary heart disease; raised apolipoprotein B-100 levels and plasma VLDL, LDL or both. 49 Familial hypertriglyceridaemia Phenotype: In the mild forms of this condition symptoms usually not evident during childhood. Environmental factors may potentiate symptoms in affected adults. Plasma triglycerides are elevated moderately. VLDL is elevated, but cholesterol is not. Lipoprotein electrophoretic patterns of type IV or occasionally type V. The severe form presents also in childhood, and is characterised by elevated triglycerides, fasting chylomicronaemia, eruptive xanthomas, recurrent abdominal pain and pancreatitis, and a type V lipoprotein pattern. Comments: Mild forms: Autosomal dominant that affects 0.2–0.3% of the population, associated with mutations in the HTGS gene. Severe forms: Autosomal dominant, resulting from mutations at the apoE gene locus that lead to defective binding with the LDL receptor and other LDL receptorrelated proteins. 50 Apolipoprotein A disorders Phenotype: Severe coronary atherosclerosis, cutaneous and tendinous xanthomas, and corneal clouding, low levels of HDL, normal LDL and cholesterol. Comment: Familial apoA-I deficiency is a rare autosomal-recessive disorder. 51 Abetalipoproteinaemia Synonym: Bassen Kornzweig disease. Phenotype: Steatorrhoea; failure to thrive; spinocerebellar ataxia, peripheral neuropathy, pigmentary retinopathy, ceroid myopathy; absent VLDL and LDL, absent apoB-48 and apoB-100 in plasma. Comment: Due to absence of a microsomal triglyceride transfer protein (MTP) involved in the assembly, processing, or secretion of apoB-containing lipoproteins. 52 Familial hypobetalipoproteinaemia Phenotype: Homozygotes: steatorrhoea; failure to thrive; spinocerebellar ataxia, peripheral neuropathy, pigmentary retinopathy, ceroid myopathy; absent VLDL and LDL; heterozygotes: sometimes ataxia or pigmentary retinopathy; low LDL and cholesterol levels. 53 Chylomicron retention disease Phenotype: Steatorrhoea, occasional neurological problems. Disorders of Glucose and Triglyceride Metabolism 85 54 Apolipoprotein C-II deficiency Phenotype: Recurrent pancreatitis; chylomicroanaemia, raised plasma triglycerides. 55 Familial dysbetalipoproteinaemia (type III hyperlipoproteinaemia) Phenotype: Cutaneous xanthomas; peripheral and coronary atherosclerosis in adults; raised plasma cholesterol and triglycerides with excess -VLDL. 56 Type V hyperlipoproteinaemia (HLP) Phenotype: Increased risk of coronary heart disease; raised plasma Lp(a). 57 Lp(a) disorders Comments: Lp(a), a variant of LDL, consists of an LDL-like molecule that has one or two copies of the glycoprotein Lp(a) attached to its apoB-100. Approximately 20% of the general population have high levels of Lp(a) that double the risk for premature atherosclerosis. 58 Tangier disease Synonym: Familial high-density lipoprotein deficiency. Phenotype: Hyperplastic orange tonsils; splenomegaly, relapsing neuropathy; low plasma cholesterol and HDL. 59 Lipoprotein lipase deficiency Phenotype: Recurrent pancreatitis; cutaneous xanthomas; hepatosplenomegaly; chylomicronaemia. 60 Familial lecithin-cholesterol acyltransferase (LCAT) deficiency Synonym: Fish eye disease (mild variant). Phenotype: LCAT deficiency: corneal opacities; anaemia; proteinuria and sometimes renal failure. Fish eye disease: corneal opacities. Little ␣1 and no pre--lipoprotein bands, unusual LDL particles with high unesterified cholesterol and phosphatidylcholine. 61 Cerebrotendinous xanthomatosis Phenotype: Tuberous and tendon xanthomas; dementia, ataxia, paraparesis; atherosclerosis; cataracts. 62 Sitosterolaemia Synonym: Phytosterolaemia. Phenotype: Tuberous and tendon xanthomas; premature coronary atherosclerosis. ESPE Classification of Paediatric Endocrine Diagnoses 86 ESPE Code Diagnosis OMIM ICD10 12 D I SO R D E R S O F BO N E M E TABO LISM , I N CLU D I N G C ALCI UM/ PH OS PHATE M E TABO LISM 12A T R A N S I E N T H Y P O C A L C A E M I A ( N E O N AT A L ) E83.5 12A.1 Early neonatal1 Prematurity 2 Asphyxia Infant of diabetic mother Perinatal stress or trauma P71.1 Late neonatal3 Disorders classified elsewhere: Conditions classified under permanent hypocalcaemia, hypomagnesaemia High milk phosphate load P71.1 12A.1a 12A.1b 12A.1c 12A.1d 12A.2 12A.2a 12A.2b P71.0, P71.1 12A.2h Parenteral nutrition Exchange transfusions Chronic alkalosis or bicarbonate treatment Maternal hypercalcaemia Maternal vitamin D deficiency Transient hypoparathyroidism 12B PERMANENT HYPOCALCAEMIA 12B.1 Genetic disorders of the calcium-sensing receptor Autosomal-dominant hypocalcaemia Autosomal-dominant hypocalcaemia with Bartter-like features 12A.2c 12A.2d 12A.2e 12A.2f 12A.2g 12B.1a 12B.1b 12B.2 12B.2a 12B.2b Hypoparathyroidism Disorders classified elsewhere: DiGeorge syndrome types 1 and 2 (14B.10) Autoimmune polyglandular syndrome type 1 (APECED syndrome) (14C.4a) X-linked hypoparathyroidism Disorders of Bone Metabolism, Including Calcium/Phosphate Metabolism P71.4 #146200 +601199 E20.9 E20.8 %307700 E20.8 87 ESPE Code Diagnosis 12B.2c 12B.2c.1 12B.2c.2 12B.2c.3 12B.2c.4 12B.2c.9 12B.2d 12B.2e 12B.2f 12B.2f.1 12B.2f.2 12B.2f.9 12B.2g 12B.2g.1 12B.2g.2 12B.2g.3 12B.2g.4 12B.2z Mitochondrial disorders Kearns-Sayre MELAS Pearson marrow-pancreas syndrome tRNA-Leu mutation Other Hypoparathyroidism, deafness and renal anomalies (Barakat syndrome) Long-chain hydroxyacyl-CoA dehydrogenase deficiency (LCHAD deficiency) [primary 11B.5d] Other familial syndromes Kenny-Caffey syndrome type 1 Sanjad-Sakati Other specified syndromes Acquired hypoparathyroidism Parathyroid surgery Isolated autoimmune hypoparathyroidism Iron overload Other specified acquired forms Idiopathic 12B.3 PTH defects (autosomal-dominant or autosomal-recessive familial isolated hypoparathyroidism) 12B.4 PTH/PTHrP receptor defects4 Pseudohypoparathyroidism type 1b Other PTH/PTHrP receptor defects, unspecified 12B.4a 12B.4z 12B.5 12B.5a 12B.5b 12B.5c 12B.5d 12B.6 12B.6a 12B.6b 12B.6c 12B.6d Post-receptor defects Classified elsewhere: Pseudohypoparathyroidism type Ia (Albright’s hereditary osteodystrophy, 14B.2) Pseudohypoparathyroidism type Ic Pseudohypoparathyroidism type II Pseudohypoparathyroidism with testotoxicosis Magnesium deficiency5 Familial primary hypomagnesaemia Familial hypomagnesaemia with hypercalciuria, nephrocalcinosis, and severe ocular involvement Isolated renal magnesium wasting Hypomagnesaemia with secondary hypocalcemia ESPE Classification of Paediatric Endocrine Diagnoses OMIM ICD10 #530000 #540000 #557000 *590050 #146255 *600890 E20.8 #244460 #241410 E20.0 #146200 E20.8 #603233 E20.1 #103580 E20.1 %203330 E20.1 #176410 E20.1 E83.4 #248250 #248190 #154020 #602014 88 ESPE Code Diagnosis 12B.6e Gitelman syndrome [primary 14B.15] 12B.7 Calciopenic rickets (see 12C.1) 12B.8 12B.8z Systemic conditions associated with hypocalcaemia Tumour lysis syndrome Renal osteodystrophy6 AIDS Other specified conditions Other conditions, unspecified 12C RICKETS 12C.1 Calciopenic rickets Nutritional (vitamin D deficiency) Malabsorption Liver disease Anticonvulsant treatment (phenobarbital, phenytoin) Renal osteodystrophy6 Calcium deficiency rickets7 Genetic Vitamin D 1␣-hydroxylase deficiency (formerly known as pseudo-vitamin D-deficiency rickets, vitamin D-dependent rickets type I) Hereditary 1,25(OH)2D-resistant rickets (formerly known as pseudo-vitamin D-deficiency rickets type II, vitamin D-dependent rickets type II, calcitriol-resistant rickets) 12B.8a 12B.8b 12B.8c 12B.8y 12C.1a 12C.1a.1 12C.1a.2 12C.1a.3 12C.1a.4 12C.1a.5 12C.1b 12C.1b.1 12C.1b.2 12C.2 12C.2a 12C.2b 12C.2b.1 12C.2b.2 12C.2b.3 12C.2b.4 12C.2c 12C.2d 12C.3 Phosphopenic rickets Classified elsewhere: McCune-Albright syndrome (14B.22) Renal tubular disorders – Fanconi renotubular syndrome (14B.13) Familial hypophosphataemic rickets X-linked hypophosphataemic rickets8 Autosomal-dominant hypophosphataemic rickets9 Hereditary hypophosphataemic rickets with hypercalciuria10 Hypophosphataemic nonrachitic bone disease Tumour-induced osteomalacia11 Decreased phosphate intake12 Hypopophosphatasia (if not associated with rickets, classify under 12E.5l) Disorders of Bone Metabolism, Including Calcium/Phosphate Metabolism OMIM ICD10 #263800 N25.0 E55.0 N25.0 #264700 E83.3 #277400 E83.3 E83.3 #307800 #193100 #241530 %146350 #146300 E83.3 #241500 89 ESPE Code Diagnosis 12D OSTEOPOROSIS 12D.1 Genetic defects Classified elsewhere: Ehlers-Danlos syndrome (14B.11) Marfan syndrome [primary 14B.20, other secondary 2A.2a] 12D.1a 12D.1b Osteogenesis imperfecta (types I–VII) OMIM ICD10 M81.9 #166200 #166210 #166220 #166240 #259420 12D.1d Homocystinuria [if tall, classify also as 2A.2c] Other genetic defects 12D.2 Chromosomal defects M82.8 12D.3 Endocrine disorders M82.0 12D.4 Iatrogenic causes M81.4 12D.5 Nutritional disorders M81.3 12D.6 Chronic diseases M82.8 12D.7 Malignancies M82.8 12D.8 Disuse M81.2 12D.9 Other specified disorders, e.g. muscle and neuromuscular disorders M81.8 12D.10 Idiopathic juvenile osteoporosis13 M81.5 12E HYPERCALCAEMIA E83.5 12E.1 Disorders of the calcium-sensing receptor Familial benign hypercalcaemia (familial hypocalciuric hypercalcaemia) Neonatal severe primary hyperparathyroidism Calcium-sensing receptor blocking antibodies 12D.1c 12E.1a 12E.1b 12E.1c ESPE Classification of Paediatric Endocrine Diagnoses +236200 #145980 #239200 90 ESPE Code Diagnosis 12E.2 12E.2a 12E.2b 12E.2c 12E.2d 12E.2e 12E.2f 12E.2y 12E.3 12E.3a 12E.4y 12E.4 12E.4a 12E.4b 12E.5 12E.5a 12E.5b 12E.5c 12E.5d 12E.5e 12E.5f 12E.5g 12E.5h 12E.5i 12E.5j 12E.5k 12E.5l 12E.5m 12E.5y 12E.5z OMIM Disorders of the parathyroid glands/PTH oversecretion Disorders classified elsewhere: Multiple endocrine neoplasia type 1 (14C.5a) Multiple endocrine neoplasia type 2a (14C.5b) Multiple endocrine neoplasia type 2b (14C.5c) Familial isolated hyperparathyroidism, type 1 Familial isolated hyperparathyroidism, type 2 (jaw tumour) Sporadic parathyroid adenomas Parathyroid carcinomas Tertiary hyperparathyroidism Other PTH abnormalities (specified) #145000 PTH/PTHrP Receptor abnormalities Metaphyseal chondrodysplasia, Jansen type Other PTH/PTHrP receptor abnormalities (specified) #156400 Abnormal vitamin D metabolism Disorders classified elsewhere: Williams-Beuren syndrome (14B.37) Idiopathic infantile hypercalcaemia14 Miscellaneous causes of hypercalcaemia in childhood Vitamin D (or vitamin D metabolite) intoxication15 Sarcoidosis, other granulomatous diseases Immobilisation16 Hypercalcaemia of malignancy Subcutaneous fat necrosis Excessive calcium supplementation Congenital lactase deficiency Disaccharidase deficiency Endocrine forms (adrenal insufficiency, severe congenital hypothyroidism, thyrotoxicosis) (primarily classified elsewhere) Down syndrome AIDS IMAGe syndrome Hypophosphatasia Other specified disorders Other disorders (unspecified) Disorders of Bone Metabolism, Including Calcium/Phosphate Metabolism ICD10 E21 #145001 E21.0 E21.0 E21.4 E21.4 E21.4 E21.4 Q78.9 #19405 E83.5 E67.3 D86.9 E83.5 E83.5 #223000 #222900 #190685 Q90 B24 300290 #241500 E83.3 91 1 Early neonatal hypocalcemia (ENH) (<72 h) Phenotype: Specific signs: tetany, generalised or focal clonic seizures, frequent twitches or jerking of limbs, hyperacusis, laringospasm, jitteriness, irritability; nonspecific signs: apnoea, tachycardia, tachypnoea, cyanosis, oedema; may be asymptomatic. Serum-Ca (–), P (+), Mg (–), PTH (+), CT (+), 25-OHD (–), 1,25(OH)2D (–). Comments: The mechanism(s) of ENH is uncertain; possible pathogenetic factors are exaggerated postnatal depression of circulating Ca levels, transient pseudohypoparathyroid-like state probably due to an immaturity of renal and bone response to PTH, and exaggerated rise in CT levels. Reduced 25-OHD and 1,25(OH)2D levels may be contributing factors. Conditions at risk to develop ENH: diabetes in the mother, pre-eclampsia, perinatal asphyxia, and prematurity. 2 Metabolic bone disease (MBD) of prematurity Synonym: Rickets of prematurity, osteopenia of prematurity. Phenotype: Asymptomatic, or hypotonia, signs of rickets and/or osteopenia, fractures of the ribs or long bones, skeletal deformities (rib cage softening, altered head shape), respiratory distress syndrome, growth delay. Serum-Ca (–/N), P (–/N), alkaline phosphatase (+/N), PTH (+/N), 25-OHD (–/N), 1,25(OH)2D (+ if Ca and P are reduced, – in case of substrate deficiency), urine-Ca (+ in phosphate depletion syndrome), urine-P (+ in secondary hyperparathyroidism, – in phosphate depletion syndrome or normal). Hormonal findings may vary on the basis of the amount of Ca and P supplied by the preterm formula, fortified human milk, or parenteral nutrition, the degree of vitamin D deficiency, and the adaptative mechanisms in response to the dietary mineral insufficiency. Comments: MBD of prematurity mainly arises from insufficient dietary minerals (Ca and P). Copper deficiency, as a result of abnormal collagen metabolism, has also been implicated in this condition. Phosphate depletion syndrome mainly occurs in preterm neonates who are fed unfortified human milk or during parenteral nutrition. 3 Late neonatal hypocalcemia (> 72 h) Phenotype: Asymptomatic, or symptoms similar to those of early neonatal hypocalcaemia. SerumCa (–), P (+), Mg (–), PTH (–/+), CT (N), 25-OHD (–), 1,25(OH)2D (–). Comments: Possible pathogenetic factors seem to be excessive dietary P load in neonates fed cow’s milk or cow’s milk based formula with Ca/P molar ratio 3–4 times that of human milk, inability of the immature kidney to excrete P efficiently, insufficient PTH response to hypocalcaemia, and materno-foetal vitamin D deficiency. Late neonatal hypocalcaemia is less frequent than early neonatal hypocalcaemia. The incidence of late neonatal hypocalcaemia is greater in full term than in preterm infants. Infants of diabetic mother and infants with transient hypoparathyroidism due to maternal hypercalcaemia secondary to primary hyperparathyroidism may be at risk for late neonatal hypocalcaemia. 4 PTH/PTHrP receptor defects Comment: A congenital defect of the PTH/PTHrP receptor is Blomstrand chondrodysplasia, a perinatally lethal condition, that does not present with hypocalcaemia. 5 Magnesium deficiency Comment: Only some conditions, such as familial primary hypomagnesaemia and hypomagnesaemia with secondary hypocalcaemia, show hypocalcaemia. ESPE Classification of Paediatric Endocrine Diagnoses 92 6 Renal osteodystrophy (ROD) Phenotype: Growth failure, bone pain, muscle weakness, skeletal deformities (scoliosis, kyphosis, genu valgum, distorsion of the thoracic cage), bone fractures (ribs, hips, vertebral bodies), extraskeletal calcifications (periarticular, vascular, subcutaneous, and visceral). Serum-P (+), Ca (–), Mg (+), alkaline phosphatase (+), PTH (+), 25-OHD (–/N), 1,25(OH)2D (–). Osteitis fibrosa (subperiosteal erosions, osteosclerosis, slipped epiphyses), rickets-like lesions. Comments: ROD is determined by the skeletal effect of secondary hyperparathyroidism associated with a reduced 1,25(OH)2D production. Reduced GFR ] decreased urinary loss of P ] mild hyperphosphataemia ] hypocalcaemia ] secondary hyperparathyroidism. As renal function continues to decline, hypocalcaemia is further aggravated by a resistance to the action of PTH on bone. In addition, the set-point for the inhibition of PTH secretion by extracellular calcium is raised in chronic renal failure, making it more difficult to turn off the secondary hyperparathyroidism. Reduced 1,25(OH)2D production derives from a decline in the functioning renal mass available to perform 1␣-hydroxylation of 25-OHD, hyperphosphataemia suppressing renal 1␣-hydroxylase activity, and metabolic acidosis. The signs and symptoms of ROD are rather nonspecific, and laboratory and radiological abnormalities often do not correspond to the severity of the clinical manifestations. 7 Calcium deficiency rickets Phenotype: Clinical features are identical to those observed in patients with vitamin D deficiency rickets. Calcium deficiency rickets usually occurs after the age of 18 months. Serum-Ca (–/N), P (–/N), alkaline phosphatase (+), PTH (N/+), 25-OHD (N/–), 1,25(OH)2D (+). Comments: Low dietary calcium intake, often exacerbated by a high oxalate and phytate content in the diet that impairs intestinal calcium absorption, is not able to ensure calcium demand for growth plate mineralisation. Reduced calcium intake ] hypocalcaemia ] secondary hyperparathyroidism ] increased 1,25(OH)2D production. Reduced 25-OH-D levels due to increased catabolism. 8 X-linked hypophosphataemic rickets (XLH) Synonyms: Hypophosphataemic vitamin D-resistant rickets. Phenotype: Growth failure with disproportionate short stature, lower limbs deformities, dental and periodontal lesions. Serum-P (–), alkaline phosphatase (+), PTH (N), 25-OHD (N), 1,25(OH)2D (– or inappropriately normal relative to hypophosphataemia), TmP/GFR (–), urine-P (+), urine-Ca (N). Osteomalacia in both cortical and trabecular bone, increase in osteoid volume, areas of hypomineralisation around osteocytes in the lamellar cortical bone. Comment: Defective tubular reabsorption of P associated with abnormal renal vitamin D metabolism caused by mutations in the PHEX (phosphate-regulating gene with homology to endopeptidases) gene, which is located on Xp22.1. 9 Autosomal-dominant hypophosphataemic rickets (ADHR) Synonym: Vitamin D-resistant rickets. Phenotype: Clinical features are similar to those observed in patients with XLH. In contrast to XLH, ADHR shows incomplete penetrance, variable age at onset (childhood to adult), and resolution of the phosphate-wasting defect in rare cases. Comments: Autosomal dominant. It is caused by mutation in a gene encoding a member of the fibroblast growth factor family (FGF23). Disorders of Bone Metabolism, Including Calcium/Phosphate Metabolism 93 10 Hereditary hypophosphataemic rickets with hypercalciuria (HHRH) Synonym: Hypercalciuric hypophosphataemic rickets. Phenotype: Stunted growth, lower limbs deformities. Serum-P (–), PTH (N), 25-OHD (N), 1,25(OH)2D (+), TmP/GFR (–), urine-P (+), urine-Ca (+). Comments: Defective tubular reabsorption of P ] hypophosphataemia ] increased 1,25(OH)2D production ] increased intestinal calcium absorption ] hypercalcaemia ] increased calcium filtered load and PTH suppression ] hypercalciuria. A significant number of asymptomatic family members showed idiopathic hypercalciuria alone without associated bone disease. Complete remission of the disease on P therapy alone. 11 Tumour rickets and osteomalacia Synonyms: Tumour-associated rickets and osteomalacia, tumour-induced rickets, oncogenic hypophosphataemic osteomalacia (OHO), oncogenic rickets and osteomalacia. Phenotype: Stunted growth, bone and muscle pain, muscle weakness, fatigue, gait disturbances, skeletal abnormalities (bowing of the lower limbs), recurrent fractures of long bones (occasionally). The majority of patients have benign or malignant tumours of mesenchymal origin of either soft tissues or bone, carcinomas of epidermal or endodermal origin; occasionally, epidermal naevus, von Recklinghausen neurofibromatosis. Serum-P (–), 25-OHD (N), 1,25(OH)2D (– or inappropriately normal relative to hypophosphatemia), TmP/GFR (–), urine-P (+). Occasionally glycinuria, glucosuria, and hyperaminoaciduria. Comments: Production of a humoral factor(s) known as ‘phosphatonin’ (FGF-23, matrix extracellular phosphoglycoprotein, frizzled-related protein 4), that may affect multiple functions of the proximal renal tubule, including P reabsorption. Complete resection of the tumour restores normophosphataemia. 12 Decreased phosphate intake Synonyms: Dietary phosphate deficiency, nutritional phosphate deficiency. Phenotype: Severe serum-P (–): red cell dysfunction (haemolytic anaemia), leukocyte dysfunction [chemotaxis (–), phagocytosis (–), bacterial killing (–)]; platelet dysfunction (thrombocytopenia); musculo-skeletal: rhabdomyolysis, reversible cardiomyopathy and myopathy; CNS: metabolic encephalopathy; metabolic acidosis, rickets/osteomalacia/osteopenia. Serum-P (–). Comments: The signs and symptoms of severe hypophosphataemia may be related to a decrease in 2,3-diphosphoglycerate and/or ATP content in tissues. Main causes: total parenteral nutrition (nutritional recovery syndrome) in which aluminium loading may be a complicating factor, low-phosphate feedings in low-birthweight infants (phosphate depletion syndrome), malabsorption. 13 Idiopathic juvenile osteoporosis Phenotype: Diffuse bone pain, mainly in the back, hips, and feet, and a characteristic gait with difficulty in walking, associated with radiological evidence of osteoporosis and fractures of the vertebrae and long bones, mostly at metaphyseal sites. Idiopathic juvenile osteoporosis may have a spontaneous recovery, usually within 3–5 years. Comments: The diagnosis of idiopathic juvenile osteoporosis is based on the exclusion of the other known causes of osteoporosis. It may be difficult to distinguish from mild forms of osteogenesis imperfecta. Bone biopsy may be useful for the correct diagnosis. ESPE Classification of Paediatric Endocrine Diagnoses 94 14 Idiopathic infantile hypercalcaemia Synonyms: Lightwood type (mild form) and Fanconi-Schlesinger type (severe forms). Phenotype: Similar to patients with Williams-Beuren syndrome; the distinction between the two syndromes remains problematic. Comment: A disorder in vitamin D metabolism with increased vitamin D sensitivity with respect to gastrointestinal transport of Ca has been postulated in some cases. Elevated serum PTHrP levels at the time of hypercalcaemia have been reported. 15 Vitamin D intoxication Synonym: Hypervitaminosis D. Phenotype: Asymptomatic or symptoms of hypercalcaemia/hypercalciuria. Excessive storage of vitamin D in adipose tissue and voluntary muscle. Serum-Ca (+), P (N/+), PTH (–), 25-OHD (+ if vitamin D or 25-OHD is ingested), 1,25(OH)2D (N), urine-Ca (+). Comment: Increased action of vitamin D metabolites on intestinal Ca absorption and Ca mobilisation from bone. 16 Immobilisation Synonyms: Immobilisation-induced hypercalciuria, immobilisation-induced hypercalcaemia and osteopenia. Phenotype: Asymptomatic or symptoms related to hypercalcaemia/hypercalciuria. Osteopenia. Serum-Ca (N/+), alkaline phosphatase (N/+), PTH (N), 1,25(OH)2D (–), urine-Ca (+), urine-P (+), GFR (–), metabolic alkalosis. Comment: Uncoupling of bone cells activity for increased osteoclastic bone resorption and decreased osteoblastic bone formation. Disorders of Bone Metabolism, Including Calcium/Phosphate Metabolism 95 ESPE Code Diagnosis 13 D I SO R D E R S O F WATE R BAL ANCE 13A D I S O R D E R S C H A R A C T E R I S E D B Y P O LY D I P S I A A N D P O LY U R I A 13A.1 Central diabetes insipidus (vasopressin deficiency)1 [primary] [secondary 6C] Genetic causes Mutations in the AVP-NPII gene2 – Familial autosomal-dominant neurohypophyseal diabetes insipidus – Autosomal-recessive neurohypophyseal diabetes insipidus Wolfram syndrome/DIDMOAD3 [primary 14B.38] [other secondary 11A.3h.4] Congenital intracranial anatomic defects Septo-optic dysplasia4 [primary 6E.1a] [other secondary 14B.30] Midline craniofacial defects Holoprosencephalic syndromes5 Agenesis of the pituitary Acquired causes (for detailed classification use codes in 6E and 6F) Neoplasms Inflammatory/infiltrative Infectious Traumatic injury Idiopathic Adipsic diabetes insipidus6 13A.1a 13A.1a.1 13A.1a.1a 13A.1a.1b 13A.1a.2 13A.1b 13A.1b.1 13a.1b.2 13a.1b.3 13a.1b.4 13A.1c 13A.1c.1 13A.1c.2 13A.1c.3 13A.1c.4 13A.1c.9 13A.1d 13A.2 13A.2a 13A.2a.1 13A.2a.2 13A.2a.3 13A.2b 13A.2b.1 13A.2b.2 13A.2b.3 Nephrogenic diabetes insipidus Genetic X-linked recessive (AVP-V2 receptor)7 Autosomal recessive (aquaporin-2)8 Autosomal dominant (aquaporin-2) Acquired Drugs, e.g. lithium, foscarnet, demeclocycline Metabolic, e.g. hyperglycaemia, hypercalcaemia, hypokalaemia, protein malnutrition Renal ESPE Classification of Paediatric Endocrine Diagnoses OMIM ICD10 E23.2 #125700 #222300 #182230 %236100 N25.1 #304800 #125800 #125800 96 ESPE Code Diagnosis OMIM ICD10 Primary polydipsia9 Psychogenic10 Dipsogenic11 Iatrogenic12 R63.1 13B DISORDERS CHAR AC TERISED BY H Y P E R N AT R A E M I A E87.0 13B.1 Disorders classified elsewhere Central diabetes insipidus (13A.1) Nephrogenic diabetes insipidus (13A.2) 13B.2 Adipsic hypernatraemia13 13B.3 Physical obstacles to drinking 13B.4 Excessive free water loss (other than diabetes insipidus) e.g. after gastroenteritis with prolonged vomiting and diarrhoea 13B.5 Excessive sodium intake Salt poisoning (child abuse) Other causes 13A.3 13A.3a 13A.3b 13A.3c 13B.5a 13B.5b 13C DISORDERS CHAR AC TERISED BY H Y P O N AT R A E M I A E87.1 13C.1 Inappropriate AVP secretion (syndrome of inappropriate antidiuretic hormone, SIADH)14 Nephrogenic syndrome of inappropriate antidiuresis (NSIAD)15 Tumours Drugs CNS disorders Non-malignant pulmonary disorders Post-operative hyponatraemia Adrenal insufficiency [primary 8A] Hypothyroidism [primary 7A] E22.2 13C.1a 13C.1b 13C.1c 13C.1d 13C.1e 13C.1f 13C.1g 13C.1h 13C.2 13C.2a 13C.2a.1 Appropriately increased secretion of vasopressin Hypovolaemic hyponatraemia from salt and water depletion Salt and water depletion Disorders of Water Balance #300539 E87.1 97 ESPE Code Diagnosis OMIM ICD10 13C.2b Primary sodium deficiency Hypervolemic hyponatraemia 13C.3 Water intoxication E87.7 13C.4 Cerebral salt wasting16 E87.1 13C.2a.2 1 Central diabetes insipidus Synonyms: Hypothalamic, neurogenic, pituitary, neurohypophyseal diabetes insipidus. Phenotype: Polyuria (exceeding 2 litres/m2/day), nocturia, enuresis, thirst, increased fluid intake, especially water. If water is restricted hypernatraemia occurs. 2 Familial central diabetes insipidus Phenotype: Polyuria and polydipsia usually after the first year of life. Undetectable AVP. Comments: Defect in the arginine vasopressin gene (AVP-neurophysin II gene). An X-linked form of neurohypophyseal diabetes insipidus has been suggested, but the evidence is weak. 3 Wolfram syndrome (DIDMOAD) Phenotype: (Partial) diabetes insipidus, gradual onset of diabetes mellitus, optic atrophy, deafness. In addition, neurogenic bladder, ataxia, psychiatric disorders. Signs outside the nervous system: hypogonadism, pigmented retinopathy, cardiomyopathy, sideroblastic anaemia, thrombocytopenia. Frequently, insulin dependence, no autoimmune phenomena. Serum AVP (–). DNA: heterogeneous with mitochondrial and nuclear mutations. Comments: Caused by mutation in the gene encoding wolframin (WFS1). Another locus for the disorder has been mapped to 4q (WFS2). 4 Septo-optic dysplasia (De Morsier Syndrome) Phenotype: Growth retardation, visual impairment, nystagmus; hypothalamic dysfunction and pituitary failure may occur. Neonatal hypoglycaemia and seizures. Developmental anomalies of the midline structures of the brain like hypoplasia of optic nerves, agenesis of septum pellucidum and agenesis of corpus callosum. Variable pituitary hormone deficiencies. Very variable phenotype. Comment: HESX1 mutations have been found in only a few cases. 5 Holoprosencephalic syndromes Phenotype: Etiologically heterogeneous entity which varies widely from cyclopia to almost no manifestation except perhaps a single middle incisor. Comments: Frequency of about 1 in 16,000 live births and about 1 in 200 spontaneous abortions. There are teratogenic causes, maternal diabetes being the most significant, giving a 200-fold increased risk. Genetic factors are indicated by familial occurrence, the occurrence of holoprosencephaly in some mendelian genetic syndromes, and the association with non-random chromosomal aberrations. One of the genetic syndromes that includes holoprosencephaly as a feature is ESPE Classification of Paediatric Endocrine Diagnoses 98 Smith-Lemli-Opitz syndrome. Several loci for holoprosencephaly have been mapped to specific chromosomal sites and the molecular defects in some cases of HPE have been identified. Holoprosencephaly-1 (HPE1) maps to 21q22.3, HPE2 is caused by a mutation in the SIX3 gene, HPE3 is caused by a mutation in the sonic hedgehog gene (SHH), HPE4 is caused by a mutation in the TGIF gene, HPE5 is caused by a mutation in the ZIC2 gene, HPE6 maps to 2q37.1, HPE7 is caused by a mutation in the PTCH1 gene, HPE8 maps to 14q13, and HPE9 is caused by a mutation in the GLI2 gene. 6 Adipsic diabetes insipidus Phenotype: Diabetes insipidus in combination with absent thirst. This can manifest itself as adipsic hypernatraemia in case of insufficient water intake (see 13B.2). 7 X-linked recessive nephrogenic diabetes insipidus Synonym: Renal diabetes insipidus. Phenotype: Polyuria, polydipsia, nocturia, compulsive drinking. In infants: failure to thrive, fever, weight loss, irritability. High serum ADH, activation of the renin-angiotensin-aldosterone system (RAAS). Comments: Nephrogenic diabetes insipidus is caused by the inability of the renal collecting ducts to absorb water in response to antidiuretic hormone (ADH), also known as arginine vasopression (AVP). Approximately 90% of patients are males with the X-linked recessive form, type I, which is caused by a mutation in the gene encoding the vasopressin V2 receptor (AVPR2). 8 Autosomal-recessive nephrogenic diabetes insipidus Synonym: Renal diabetes insipidus. Phenotype: Polyuria, polydipsia, nocturia, compulsive drinking. In infants: failure to thrive, weight loss, fever. High serum ADH, activation of the renin-angiotensin-aldosterone system (RAAS). Comments: Nephrogenic diabetes insipidus is caused by the inability of the renal collecting ducts to absorb water in response to antidiuretic hormone (ADH), also known as arginine vasopressin (AVP). 10% of patients have the autosomal form, type II, caused by mutation in the AQP2 gene. Both autosomal dominant and autosomal recessive forms have been reported. 9 Primary polydipsia Synonym: Primary polyuria. Phenotype: Excessive water drinking, resulting in decrease of plasma osmolality. Normalisation of renal concentrating capacity by stepwise reduction of water intake. Hypernatraemia is never seen. Therapy with dDAVP may cause water intoxication to develop rapidly. 10 Psychogenic polydipsia Phenotype: Can occur as part of a general cognitive defect associated with schizophrenia or other psychiatric disorder or compulsive water drinking. 11 Dipsogenic polydipsia Phenotype: Increased water consumption is due to an increase in thirst, e.g. in diseases involving the hypothalamus. . 12 Iatrogenic polydipsia Phenotype: Primary polydipsia can also be prompted by incorrect advice or incorrect understanding of advice offered by physicians, etc. Disorders of Water Balance 99 13 Adipsic hypernatraemia Phenotype: Primary adipsia is usually caused by lesions in the anterior hypothalamus. The water intake associated with a normal diet is insufficient to match obligate renal, bowel, and insensible water losses, and absent thirst can lead to hypernatraemic dehydration. 14 Inappropriate AVP secretion (syndrome of inappropriate antidiuretic hormone, SIADH) Phenotype: Euvolaemic hyponatraemia, concentrated urine, sodium concentration >20 mmol/l, low serum uric acid and urea concentration. Can be caused by ADH-producing tumours, pulmonary and CNS disorders of different origin, drugs, and others. Inappropriately high serum ADH, elevated serum ANF. Comments: The syndrome of inappropriate antidiuretic hormone secretion (SIADH) is a common cause of hyponatraemia. The syndrome manifests as an inability to excrete a free water load, with inappropriately concentrated urine and resultant hyponatraemia, hypo-osmolality, and natriuresis. SIADH occurs in a setting of normal blood volume, without evidence of renal disease or deficiency of thyroxine or cortisol. Although usually transient, SIADH may be chronic; it is often associated with drug use or a lesion in the central nervous system or lung. 15 Nephrogenic syndrome of inappropriate diuresis (NSIAD) Comments: Nephrogenic syndrome of inappropriate antidiuresis (NSIAD) is characterised by a clinical picture similar to SIADH, but is associated with undetectable levels of AVP. The disorder is caused by gain-of-function mutations in the gene encoding the vasopression V2 receptor (AVPR2). Constitutive activation of the receptor results in antidiuresis. 16 Cerebral salt wasting Comment: Following CNS injury, a syndrome of hyponatraemia associated with increased urine sodium concentration, increased urine volume, and volume depletion known as salt wasting can develop. ESPE Classification of Paediatric Endocrine Diagnoses 100 ESPE Code Diagnosis OMIM ICD10 14 SYN D ROM ES WITH E N DOCR I NE FE ATUR ES 1 14A CHROMOSOMAL ABNORMALITIES (deletions or duplications of complete or half chromosomes) 14A.1 Chromosome 18q deletion syndrome2 If associated with short stature: also classify as 1A.1a #601808 Q93.5 14A.2 Down syndrome, trisomy 213 If associated with any other disorder, also classify there For example: If associated with short stature: also classify as 1A.1a If associated with obesity: also classify as 5B.3a If associated with autoimmune hypothyroidism: also classify as 7A.3b If associated with diabetes mellitus: also classify as 11A.3h.1 #190685 Q90 14A.3 Klinefelter syndrome (47,XXY)4 If associated with tall stature: also classify as 2A.1a If associated with obesity: also classify as 5B.3a If associated with hypogonadism: also classify as 9A.0 If associated with diabetes mellitus: also classify as 11A.3h.2 Q98.0 14A.4 Mixed gonadal dysgenesis (45,X/46,XY)5 If associated with disorder of sex development, primarily classify as 4A.1 If associated with short stature: also classify as 1A.1a Q99.9 14A.5 Turner syndrome (Ulrich-Turner syndrome) (45,X or variants)6 If associated with short stature: also classify as 1A.1a If associated with obesity: also classify as 5B.3a If associated with gonadal dysgenesis: also classify as 10A.1a If associated with diabetes mellitus: also classify as 11A.3h.3 Q96.9 14A.6 XYY syndrome7 If associated with tall stature: also classify as 2A.1a Q98.5 Syndromes with Endocrine Features 101 ESPE Code Diagnosis OMIM ICD10 14B C O N G E N I TA L DY S M O R P H I C S Y N D R O M E S 14B.1 Aarskog-Scott syndrome8 If associated with short stature: also classify as 1A.1a If associated with shawl scrotum: also classify as 9F.2 100050 Q87.1 14B.2 Albright’s hereditary osteodystrophy, pseudohypoparathyroidism9 If associated with hypocalcaemia:also classify as 12B.5a If associated with short stature: also classify as 1A.3y If associated with obesity: also classify as 5B.2a #103580 E20.1 14B.3 Alström syndrome10 If associated with obesity: also classify as 5A.2a If associated with diabetes mellitus: also classify as 11A.3h.11 #203800 Q87.8 14B.4 Bannayan-Riley-Ruvalcaba syndrome11 If associated with tall stature: also classify as 2A.3a #153480 Q87.3 14B.5 Beckwith-Wiedemann syndrome12 If associated with tall length/stature: also classify as 2A.4a If associated with hypoglycaemia: also classify as 11B.1a.5 #130650 Q87.3 14B.6 Bloom syndrome13 If associated with short stature: also classify as 1A.1a #210900 Q82.8 14B.7 Carpenter syndrome14 If associated with obesity: also classify as 5B.2a %201000 Q87.0 14B.8 Cohen syndrome15 If associated with obesity: also classify as 5B.2a #216550 Q87.8 14B.9 Cornelia de Lange syndrome16 If associated with short stature: also classify as 1A.1a #122470 Q87.1 #300590 #610759 14B.10 DiGeorge/velocardiofacial syndrome syndrome (part of CATCH 22 complex)17 If associated with short stature: also classify as 1A.1a If associated with hypocalcaemia: also classify as 12B.2a ESPE Classification of Paediatric Endocrine Diagnoses #188400 Q87.1 102 ESPE Code Diagnosis 14B.11 Ehlers-Danlos syndrome18 If associated with osteoporosis: also classify as 12D.1a OMIM ICD10 #130000 Q79.6 (to 80) #225400 %229200 %305200 14B.12 Elejalde syndrome19 If associated with tall stature: also classify as 2A.3a 200995 Q87.3 14B.13 Fanconi renotubular syndrome20 If associated with short stature: also classify as 1B.3a.0 If associated with rickets: also classify as 12C.2a %134600 E72.0 14B.14 Fragile X syndrome21 If associated with tall stature: also classify as 2A.3a #300624 Q99.2 14B.15 Gitelman syndrome22 If associated with hypocalcaemia: classify as 12B.6e #263800 14B.16 Kallmann syndrome23 Primarily classified as 6A.3a.1 If associated with tall stature: also classify as 2B.6a +308700 14B.17 Klippel-Trenaunay-Weber syndrome24 If associated with tall stature: also classify as 2A.4a %149000 Q87.2 14B.18 Laurence-Moon-Bardet-Biedl syndrome25 If associated with obesity: also classify as 5B.2a If associated with hypogonadism: also classify as 9A.0 If associated with diabetes mellitus: also classify as 11A.3h.7 245800 Q87.8 14B.19 Leri-Weill dyschondrosteosis26 If associated with short stature: also classify as 1A.1a #127300 Q77.8 14B.20 Marfan syndrome27 If associated with tall stature: also classify as 2A.2a If associated with osteoporosis: also classify as 12D.1a #154700 Q87.4 14B.21 Marshall-Smith syndrome28 If associated with tall stature: also classify as 2A.3a 602535 Syndromes with Endocrine Features E23.0 #147950 #244200 #209900 103 ESPE Code Diagnosis OMIM ICD10 14B.22 McCune-Albright syndrome29 #174800 If associated with tall stature: also classify as 2B.1b If associated with pseudoprecocious puberty: also classify as 3A.2c.0 If associated with hyperthyroidism: also classify as 7B.1d.1 If associated with phosphogenic rickets: also classify as 12C.2a Q87.1 14B.23 Nevo syndrome30 If associated with tall stature: also classify as 2A.3a #601451 E87.3 14B.24 Noonan syndrome31 If associated with short stature: also classify as 1A.1a If associated with hypogonadism: also classify as 9A.0 #163950 Q87.1 14B.25 Prader-Willi(-Labhart) syndrome32 If associated with short stature: also classify as 1A.1a If associated with obesity: also classify as 5B.2a If associated with hypogonadotrophic hypogonadism: also classify as 6A.3d If associated with hypothalamic-pituitary disorders: also classify as 6E.2a If associated with diabetes mellitus: also classify as 11A.3h.10 #176270 Q87.1 14B.26 Proteus syndrome33 If associated with tall stature: also classify as 2A.4a #176920 Q87.3 14B.27 Von Recklinghausen’s disease (neurofibromatosis type 1)34 If associated with short stature: also classify as 1A.1a If associated with precocious puberty: also classify as 3A.1a and 6F.1a.5 +162200 Q85.0 14B.28 Rieger syndrome35 If associated with growth hormone deficiency: also classify as 1B.3a.0 If associated with ACTH deficiency: also classify as 6A.1 If associated with TSH deficiency: also classify as 6A.2 If associated with gonadotrophin deficiency: also classify as 6A.3d #180500 Q87.1 14B.29 Rubinstein-Taybi syndrome36 If associated with short stature: also classify as 1A.1a #180849 Q87.2 ESPE Classification of Paediatric Endocrine Diagnoses 104 ESPE Code Diagnosis OMIM ICD10 14B.30 Septo-optic dysplasia37 Primarily classified as 6E.1a If associated with short stature: also classify as 1B.3a.3 If associated with diabetes insipidus: also classify as 13A.1b.1 #182230 Q04.4 14B.31 Silver-Russell syndrome38 If associated with short stature: also classify as 1A.1a If associated with precocious or delayed puberty: also classify according to the codes in chapter 3 %180860 Q87.1 14B.32 Simpson-Golabi-Behmel syndrome (mutations in GPC3)39 If associated with tall stature: also classify as 2A.3a #312870 Q87.3 14B.33 Smith-Lemli-Opitz syndrome40 If associated with a disorder of sex development: also classify as 4B.2a If associated with low cholesterol level: also classify as 11C.4a #270400 Q87.1 14B.34 Sotos syndrome41 If associated with tall stature: also classify as 2A.3a #117550 Q87.3 14B.35 Steinert’s disease, myotonic dystrophy 142 If associated with precocious puberty: also classify as 3A.1y If associated with delayed puberty: also classify as 3D.0 If associated with hypothyroidism: also classify as 7A.3y If associated with hyperthyroidism: also classify as 7B.2a If associated with adrenal insufficiency: also classify as 8A.3a If associated with hypogonadism: also classify as 9A.0 If associated with diabetes mellitus: also classify as 11A.3h.8 #160900 G71.1 14B.36 Weaver syndrome43 If associated with tall stature: also classify as 2A.3a #277590 Q87.3 14B.37 Williams-Beuren syndrome44 If associated with short stature: also classify as 1A.1a If associated with hypercalcaemia: also classify as 12E.4a #194050 Q87.1 14B.38 Wolfram syndrome (DIDMOAD)45 If associated with diabetes mellitus: also classify as 11A.3h.4 If associated with diabetes insipidus: also classify as 13A.1a.2 #222300 Syndromes with Endocrine Features E10 E23.2 105 ESPE Code Diagnosis 14C N O N - DYSM O R PH I C SY N D RO M E S 14C.1 Cushing syndrome46 Primarily classified as 8C.1 If associated with short stature: also classify as 1B.5a If associated with diabetes mellitus: also classify as 11A.3d.2 14C.2 Mauriac syndrome47 Primarily classified as 11A If associated with short stature: also classify as 1B.5a If associated with obesity: also classify as 5C.0 14C.3 Polycystic ovary syndrome48 Primarily classified as 10A.2a If associated with hirsutism: also classify as 3C.2a If associated with obesity: also classify as 5C.0 14C.4 Autoimmune polyglandular syndromes Autoimmune polyglandular syndrome type 1 (APECED)49 If associated with autoimmune thyroiditis: also classify as 7A.3b If associated with autoimmune adrenalitis: also classify as 8A.3a If associated with autoimmune hypergonadotrophic hypogonadism: also classify as 9A.2a or 10A.1a If associated with type 1 diabetes mellitus: also classify as 11A.1a If associated with hypoparathyroidism: also classify as 12B.2a Autoimmune polyglandular syndrome type 250 If associated with autoimmune thyroiditis: also classify as 7A.3b If associated with autoimmune adrenalitis: also classify as 8A.3a If associated with autoimmune hypergonadotrophic hypogonadism: also classify as 9A.2a or 10A.1a If associated with type 1 diabetes mellitus: also classify as 11A.1a If associated with hypoparathyroidism: also classify as 12B.2a Autoimmune polyglandular syndrome type 351 If associated with autoimmune thyroiditis: also classify as 7A.3b If associated with autoimmune hypergonadotrophic hypogonadism: also classify as 9A.2a or 10A.1a If associated with type 1 diabetes mellitus: also classify as 11A.1a If associated with hypoparathyroidism: also classify as 12B.2a Immune dysregulation, polyendocrinopathy, and enteropathy (X-linked) syndrome (IPEX)52 Autoimmune lymphoproliferative syndrome (ALPS)53 14C.4a 14C.4b 14C.4c 14C.4d 14C.4e ESPE Classification of Paediatric Endocrine Diagnoses OMIM ICD10 E24 #184700 E28.2 M35.9 #240300 M35.9 269200 M35.9 M35.9 #304790 M35.9 #601859 M35.9 106 ESPE Code Diagnosis OMIM ICD10 %147920 M35.9 14C.4f Kabuki make-up syndrome54 If associated with short stature: also classify as 1A.1a If associated with growth hormone deficiency: also classify as 1B.3a.0 If associated with Hashimoto’s thyroiditis: also classify as 7A.3b If associated with primary ovarian dysfunction: also classify as 10A.1c.8 14C.5 Multiple endocrine neoplasia (MEN) syndromes Multiple endocrine neoplasia 155 +131100 If associated with hyperparathyroidism: also classify as 12E.2a If associated with prolactinoma: also classify as 6B.3a If associated with growth hormone-secreting tumour: also classify as 2B.1b If associated with insulinoma: also classify as 11B.1b.7 If associated with adrenal cortical tumour (carcinoma): also classify as 8C.1b.4 If associated with thyroid adenoma: also classify as 7D.1 MEN2A56 #171400 If associated with medullary thyroid carcinoma: also classify as 7D.2c.2 If associated with phaeochromocytoma: also classify as 8D.2a If associated with hyperparathyroidism: also classify as 12E.2a MEN2B57 #162300 If associated with medullary thyroid carcinoma: also classify as 7D.2c.3 If associated with phaeochromocytoma: also classify as 8D.2a 14C.5a 14C.5b 14C.5c M8360/1 M8360/1 M8360/1 1 Syndromes with endocrine features Comments: Endocrine syndromes without significant other features are not classified in this chapter, but only in the pertinent organ-specific chapters. Information in this chapter is mainly derived from the OMIM database, in which more detailed and updated information can be found. 2 Chromosome 18q deletion syndrome Synonyms: Chromosome 18q- syndrome, 18q- syndrome. Phenotype: The phenotype is highly variable, but is characterised by mental retardation, short stature, hypotonia, hearing impairment, and foot deformities. Tapered digits and wide mouth have been described. Comments: Its basis is assumed to be in haploinsufficiency of multiple genes. It is a terminal deficiency or macrodeletion syndrome. Syndromes with Endocrine Features 107 3 Down syndrome Synonyms: Trisomy 21, trisomy-21-syndrome, mongolism. Phenotype: (1) Dysmorphic features (brachycephaly, Brushfield spots, simian crease, gap between 1st and 2nd toe, high arched palate, strabismus, broad short neck, small teeth, short broad hands, hypoplasia of the middle phalange V); (2) psycho-motor (mental retardation, hypotonia); (3) short stature (approximately 100%); (4) cardiac anomalies (40% atrio-ventricularis communis); (5) infertility (100%); (6) frequent obesity, leukaemia (1%), urinary tract malformations and hypothyroidism. Comments: Trisomy of chromosome 21, additional (maternal) chromosome free or translocated (3%) mosaicism (2%). Full clinical picture in trisomy 21p22. Mostly sporadic except for maternal translocation. Incidence 1:650, increasing with age of mother. 4 Klinefelter syndrome (47,XXY) Phenotype: From childhood: normal/tall stature in comparison to parental heights, with or without mild learning difficulties and behavioural problems, small head circumference, testes normal/small. In adolescence: obesity, long legs, prolonged growth resulting in tall stature, gynaecomastia, female pubic hair distribution, testes small and firm. Basal LH (+), FSH (+), GnRH response (+) by mid-puberty. T response (–/N) to hCG. Partial Leydig cell failure. Adulthood: Infertility + gynaecomastia (15%) with weak facial hair growth, obesity. Progressive loss of sperm cells, Sertoli cells. Hyalinisation of seminiferous tubules with relative preservation of Leydig cells. Azoospermia/oligospermia. Karyotype: 47,XXY or 46XY/47XXY or mosaicism. Comments: 1 in 600 male births. Testosterone replacement during adolescence may lessen disproportion and tall stature. Mosaicism may occur, and results in a milder clinical picture. 5 Mixed gonadal dysgenesis (MGD) Phenotype: Variable degree of impaired masculinisation (4A.1), from almost female (in case of complete female phenotype classify as Turner syndrome (14A.5)) to normal male genitalia (14A.4). Hypospadias, cryptorchidism may be present. At puberty FSH and LH are usually elevated and testosterone decreased. Secondary to sex chromosomal mosaicism in a male, the gonads have become dysgenetic in combinations ovary/testis, or testis/streak. Sometimes a tumour is found instead of a gonad. Comments: The term MGD can be used when the above chromosomal mosaicism (or, in rare cases, 45,X/46,XY/47,XYY) is found, even if gonadal dysgenesis has not been proven anatomically. 6 Turner syndrome (TS) or Ullrich-Turner syndrome (UTS) Phenotype: Female appearance, short stature (95%), gonadal dysgenesis (90%), dysmorphic stigmata (pterygium colli, cubitus valgus), coarctation of the aorta, renal and urinary tract malformation. Great variability in physical appearance. In early infancy and from 10 years LH and FSH (+) and E2 (–). Karyotype: loss or abnormality of one X chromosome or mosaicism (typical karyotype: 45,X). Comment: Approximately 1:2,000 (female births). 7 XYY syndrome Phenotype: Often no specific symptoms, sometimes behavioural problems. Adulthood: FSH (N/+), reduced fertility. Histology: quantitatively reduced spermatogenesis. Comment: 1/900 newborn males. ESPE Classification of Paediatric Endocrine Diagnoses 108 8 Aarskog-Scott syndrome Phenotype: Short stature, hypertelorism, shawl scrotum, delayed puberty, cryptorchidism, stretchable skin. Comment: X-linked recessive inheritance or autosomal dominant. 9 Albright’s hereditary osteodystrophy (AHO), pseudohypoparathyroidism Synonyms: Albright syndrome, hereditary osteodystrophy, pseudohypoparathyroidism type Ia (PHP Ia). Phenotype: Short stature, obesity, round facies, subcutaneous ossifications, brachydactyly, short IV metacarpal, short thick neck, and other skeletal anomalies. Some patients have mental retardation. AHO is often associated with pseudohypoparathyroidism, hypocalcaemia, tetany, hyperphosphataemia and PTH (+) (PHP Ia). Patients with pseudopseudohypoparathyroidism have normal calcium metabolism and PTH levels with isolated AHO. T4 and T3 (N/–), TSH (N/+), parathyroids normal or hyperplastic, calcification of basal ganglia. Comments: Heterogeneous group of disorders whose common feature is resistance to parathyroid hormone (PTH). It is generally classified as types Ia, Ib, Ic, and II according to different phenotypes and pathogenesis. Albright hereditary osteodystrophy (AHO), pseudohypoparathyroidism type Ia (PHP Ia), and pseudopseudohypoparathyroidism (PPHP) are caused by mutations, including imprinting defects, in the GNAS1 gene. Pseudohypoparathyroidism should not be confused with polyostotic fibrous dysplasia, to which Albright’s name is also attached. 10 Alström syndrome Phenotype: Although this disorder bears many similarities (retinitis pigmentosa, progressive nerve deafness, obesity, and diabetes mellitus) to the Bardet-Biedl syndrome there is no mental defect, polydactyly, or hypogonadism. The retinal lesion causes nystagmus and early loss of central vision in contrast to loss of peripheral vision first, as in other pigmentary retinopathies. Hypogonadotrophic hypogonadism (FSH (–), LH (–)), insulin resistance (insulin (+)), acanthosis nigricans, glomerulosclerotic renal disease. Comment: Caused by mutation in the ALMS1 gene. 11 Bannayan-Riley-Ruvalcaba syndrome Synonyms: Bannayan-Zonana, Ruvalcaba-Myhre, Riley-Smith, Bannayan-Riley-Ruvalcaba. Phenotype: Prenatal overgrowth with large birth weight and length. Developmental delay with hypotonia and macrocephaly with frontal bossing. Downslanting palpable fissures. Pigmented macules on penis. Mesodermal hamartomas with angiolipomas/lipomas in GI tract, cranium, bone. Lipid storage myopathy. Comment: Caused by mutations in the PTEN gene. 12 Beckwith-Wiedemann syndrome (BWS) Synonym: Beckwith syndrome. Phenotype: Macrosomia particularly affecting tongue and kidneys, exomphalos, ear lobe creases and pits. Pancreatic hyperplasia with hyperinsulinism. Hemihypertrophy and predisposition to tumours, especially Wilms’ (7%). Large muscle mass and thick subcutaneous tissue with organomegaly. Transient hyperinsulinism resulting in hypoglycaemia. Syndromes with Endocrine Features 109 Comments: Genetic lesion probably causes imbalance between maternally imprinted IGF-2 growth enhancer gene and paternally imprinted H19 growth suppressor gene, leading to foetal overgrowth. Affected infants require clinical, ultrasound, and alpha-foetoprotein monitoring until growth is completed to pre-empt tumour development, particularly Wilms’. 13 Bloom syndrome Phenotype: Bloom syndrome is an autosomal-recessive disorder characterised by proportionate pre- and postnatal growth deficiency; sun-sensitive, telangiectatic, hypo- and hyperpigmented skin; predisposition to malignancy; and chromosomal instability. Comment: Caused by mutations in the gene encoding DNA helicase RecQ protein-like-3. 14 Carpenter syndrome Synonym: Acrocephalopolysyndactyly. Phenotype: Acrocephaly, peculiar facies, brachysyndactyly of the fingers, preaxial polydactyly and syndactyly of the toes, hypogenitalism, obesity and mental retardation, short stature. LH (–), FSH (–). 15 Cohen syndrome Phenotype: Nonprogressive mild to severe psychomotor retardation, motor clumsiness, microcephaly, characteristic facial features, childhood hypotonia and joint laxity, progressive retinochoroidal dystrophy, myopia, intermittent isolated neutropenia, and a cheerful disposition. Characteristic facial features include high-arched or wave-shaped eyelids, a short philtrum, thick hair, and low hairline. Comments: Rare autosomal-recessive disorders that are overrepresented in the Finnish population. Some patients with Cohen syndrome have mutations in the COH1 gene. 16 Cornelia de Lange syndrome Phenotype: The de Lange syndrome is recognised on the basis of characteristic facies (low anterior hairline, synophrys, anteverted nares, maxillary prognathism, long philtrum, ‘carp’ mouth) in association with prenatal and postnatal growth retardation, mental retardation and, in many cases, upper limb anomalies. Comments: Can be caused by mutation in the NIPBL gene, which encodes a component of the cohesin complex. About half of the cases of CDLS are due to mutations in this gene. An X-linked form of the disorder (CDLS2) can be caused by mutation in the SMC1L1 gene, which also encodes a component of the cohesin complex. A mild variant of Cornelia de Lange syndrome (CDLS3) has been related to mutation in the SMC3 gene, which encodes another component of the cohesin complex. 17 DiGeorge syndrome Synonym: 22q11.2 deletion syndrome, hypoplasia of thymus and parathyroids, third and fourth pharyngeal pouch syndrome, takao vcf syndrome, catch22. Phenotype: DiGeorge syndrome (DGS) comprises hypocalcaemia arising from parathyroid hypoplasia, thymic hypoplasia, and outflow tract defects of the heart. Disturbance of cervical neural crest migration into the derivatives of the pharyngeal arches and pouches can account for the phenotype. Most cases result from a deletion of chromosome 22q11.2 (the DiGeorge syndrome chromosome region, or DGCR). Several genes are lost including the putative transcription factor TUPLE1 ESPE Classification of Paediatric Endocrine Diagnoses 110 which is expressed in the appropriate distribution. This deletion may present with a variety of phenotypes: Shprintzen or velocardiofacial syndrome; conotruncal anomaly face (or Takao syndrome); and isolated outflow tract defects of the heart including tetralogy of Fallot, truncus arteriosus, and interrupted aortic arch. A collective acronym CATCH22 has been proposed for these differing presentations. A small number of cases of DGS have defects in other chromosomes, notably 10p13. Comments: DiGeorge syndrome is caused by a 1.5- to 3.0-Mb hemizygous deletion of chromosome 22q11.2. Haploinsufficiency of the TBX1 gene in particular is responsible for most of the physical malformations. There is evidence that point mutations in the TBX1 gene can also cause the disorder. 18 Ehlers-Danlos syndrome Phenotype: The main features of classic Ehlers-Danlos syndrome, which includes EDS I and EDS II, are loose-jointedness and fragile, bruisable skin that heals with peculiar ‘cigarette-paper’ scars. Most persons are born prematurely due to premature rupture of foetal membranes. A ‘spontaneous’ carotid-cavernous fistula and rupture of large vessels, hiatus hernia, spontaneous rupture of the bowel, diverticula of the bowel and retinal detachment have been reported. Comments: According to the classification used by McKusick, 11 types of EDS were distinguished. In a later classification major and minor diagnostic criteria were defined for each type and complemented whenever possible with laboratory findings. Six main descriptive types were substituted for earlier types numbered with Roman numerals: classic type (EDS I and II), hypermobility type (EDS III), vascular type (EDS IV), kyphoscoliosis type (EDS VI), arthrochalasia type (EDS VIIA and VIIB), and dermatosparaxis type (EDS VIIC). Six other forms were listed, including a category of ‘unspecified forms. At least some cases of the syndrome are caused by mutation in the collagen alpha-1(V) gene (COL5A1), the collagen alpha-2(V) gene (COL5A2), or the collagen alpha-1(I) gene (COL1A1). 19 Elejalde syndrome Synonyms: Acrocephalopolydactylous dysplasia, acrocephalopolydactyly renal dysplasia syndrome. Phenotype: Excessive birth weight, a swollen globular body with thick skin, apparently short limbs, polydactyly, craniosynostosis with acrocephaly, omphalocele, and abnormal face. At autopsy abdominal organomegaly, ascites, and cystic renal dysplasia, with excessive amounts of connective tissue and perivascular proliferation of nerve fibers in many organs. Comment: Not to be confused with Elejalde’s disease (neuroectodermal melanolysosomal disease). 20 Fanconi’s syndrome (FS) Phenotype: Anorexia, vomiting, polydipsia, polyuria, constipation, unexplained fever (may reflect episodic dehydratation), delayed psychomotor development, growth failure, bone disease (rickets resistant to doses of vitamin D that are ordinarily adequate for treatment of vitamin D deficiency rickets, bone pain, osteopenia, fractures), muscle weakness and episodic paralysis due to severe hypokalaemia, nephrolithiasis, nephrocalcinosis, symptoms related to the cause of the disorder. Excessive renal excretion of Na, K, P, Ca, glucose, aminoacids, bicarbonate, uric acid, citrate and other organic acid, and low-molecular-weight proteins (<50.000 Da), chronic hyperchloraemic metabolic acidosis; serum-P (–), K (–), Ca (N/–), alkaline phosphatase (+ if rickets is present), 25-OHD (N/– if hepatic damage is present), 1,25(OH)2D (–/N). Comments: Generalised defect in renal proximal tubule transport capacity; vitamin D resistance may be due to an impaired 1,25(OH)2D production by abnormal proximal tubular cells in the presence of Syndromes with Endocrine Features 111 metabolic acidosis. Most commonly it is idiopathic, or related to genetically transmitted inborn errors of metabolism (cystinosis, fructose intolerance, galactosaemia, glycogenosis, Lowe syndrome, tyrosinaemia, Wilson’s disease), or acquired. FS may be associated with renal tubular acidosis. 21 Fragile X syndrome Phenotype: Moderate to severe mental retardation (possibly worse with age); particularly delayed speech development; behavioural changes; seizures. Large forehead and head (circumference >97th centile), prominent chin, long nose and ears; increased birth weight, tall stature in childhood but adult height tends to shortness; macro-orchidism in males (15% prepubertally, 80% by adulthood – up to 120 ml); increased risk of mitral valve prolapse, aortic root dilatation and hernias; soft skin; joint hypermobility. Testes are usually histologically and functionally normal; increased size may be due to interstitial oedema or connective tissue; normal size penis. Generally normal gonadal function, normal GH axis, no precocious puberty. Chromosomal analysis: Xq27 folate-sensitive fragile site in 5–20% of cells. Comments: Basically inherited as an X-linked recessive disorder, i.e. a female may carry the trait and pass it on to her sons; only 50% of obligatory female carriers demonstrate fragile sites. 1/1,000– 1,500 males; 5% of ‘autistic’ males; second commonest cause of mental retardation (after Down syndrome); up to one third of female heterozygotes may show some manifestation of the disorder (<10% of all female mild mental retardation). 22 Gitelman syndrome Phenotype: It has been proposed that Bartter syndrome, defined generically as an autosomal-recessive disorder featuring hypokalaemic metabolic alkalosis with salt wasting, is a heterogeneous entity with at least 2 subsets, Gitelman syndrome and ‘true Bartter syndrome’. Gitelman syndrome refers to the numerically predominant subset of patients with hypokalaemic alkalosis in conjunction with hypocalciuria and hypomagnesaemia, while true Bartter syndrome refers to patients with normal or hypercalciuria and typically normal magnesium levels. True Bartter patients usually present under the age of 5 years with signs of vascular volume depletion, while Gitelman syndrome patients typically present at older ages without overt hypovolaemia. Comment: The Gitelman variant of Bartter syndrome is caused by mutation in the thiazide-sensitive Na-Cl cotransporter (SLC12A3). 23 Isolated hypogonadotrophic hypogonadism and anosmia (Kallmann syndrome) Phenotype: Clinically and genetically heterogeneous disorder with a wide spectrum of reproductive function and anosmia. Absence of puberty, anosmia. Histologically immature gonads. LH (–), FSH (–), T (–), inhibin-B (–). Hypoplasia of the bulbus olfactorius in MRI. In severe forms hypoplastic genitalia. Comments: Lack of GnRH neurones. X-linked mutations in KAL-1 gene (encodes anosmin-1, a protein involved in olfactory and neuronal migration). Autosomal-dominant loss of function mutations in FGFR1 gene. Anosmia occurs in all KAL-1 mutations but only a small percentage of FGFR-1 mutations. 24 Klippel-Trenaunay-Weber syndrome Phenotype: Triad of (1) cutaneous vascular naevus; (2) varicosity over trunk or limbs (usually lower) in asymmetrical distribution; (3) hypertrophy of soft tissue and bone. Naevus formation and bone hypertrophy do not always overlap. Occasional neurological involvement with meningocortical angiomata. ESPE Classification of Paediatric Endocrine Diagnoses 112 Comments: Somatic mutation affecting factor involved in embryonic vascular development. Usually sporadic but increased familial incidence of hemi-hypertrophy in some families. 25 Laurence-Moon-Bardet-Biedl syndrome Synonyms: Laurence-Moon syndrome; Bardet-Biedl syndrome (it is assumed that these may be two separate entities). Phenotype: Retinal dystrophy (retinitis pigmentosa), polydactyly or syndactyly, increased appetite, mental retardation, mild obesity, hypogonadism, defective pubertal development. Renal abnormalities are frequent in the Bardet-Biedl syndrome. FSH (N, –), LH (N, –), T (–). Comment: Mutations have been found in genes at the BBS1, BBS2, BBS3, BBS4, BBS5, BBS6, BBS7, BBS8, BBS9, BBS10, BBS11, and BBS12 loci. 26 Leri-Weill dyschondrosteosis Synonym: Leri-Weill syndrome. Phenotype: Mesomelic (shortening of forearms and lower legs) short stature, with Madelung’s deformity. Heterogeneity of the clinical expression, e.g. short fourth metacarpals, scoliosis, exostoses. Adult height of males 156–171 cm, females 135–164 cm. Comments: DNA: haploinsufficiency of SHOX. A complete (homozygous) defect of SHOX causes Langer mesomelic dwarfism. Heterozygous SHOX mutations or deletions have been described in 2.5% of short children previously labeled idiopathic short stature. 27 Marfan syndrome Phenotype: A heritable disorder of fibrous connective tissue, Marfan syndrome shows striking pleiotropism and clinical variability. The cardinal features occur in 3 systems – skeletal, ocular, and cardiovascular. Increased height, disproportionately long limbs and digits, anterior chest deformity, mild to moderate joint laxity, vertebral column deformity (scoliosis and thoracic lordosis), and a narrow, highly arched palate with crowding of the teeth are frequent skeletal features. Tall stature with characteristic phenotype – arachnodactyly, long face, high palate. Hyperextensible joints with hypotonia, kyphoscoliosis, pes planus. Upward lens subluxation, myopia, retinal detachment. Aortic root dilatation leading to rupture, mitral valve prolapse. Aortic histology shows swelling and fragmentation of elastic fibres. Microfibrillopathy with abnormal and reduced microfibril production in connective tissues. Comments: All cases of the true Marfan syndrome appear to be due to mutation in the fibrillin-1 gene (FBN1) gene. Incidence: 1 in 20,000. Variable phenotype reflects different mutations. Cardiac ultrasonography is a useful screening test in children with unexplained tall stature/mildly marfanoid habitus. NB: Several rare syndromes described with marfanoid habitus and other features (e.g. mental retardation, craniosynostosis, X linked inheritance). 28 Marshall-Smith syndrome Phenotype: Prenatal onset acceleration in linear growth with failure to thrive, developmental delay, unusual facies, including prominent forehead, shallow orbits, blue sclerae, depressed nasal bridge, and micrognathia. Respiratory difficulties causing early death from pneumonia. Radiology: accelerated skeletal maturation, broad proximal and middle, with narrow distal, phalanges. Cerebral anomalies (macrogyria, cerebral atrophy, absent corpus collosum, cerebellar hypoplasia). Syndromes with Endocrine Features 113 29 McCune-Albright syndrome (MAS) Phenotype: The classic triad is café-au-lait spots, polyostotic fibrous dysplasia and gonadotrophin independent precocious puberty. Also thyrotoxicosis, pituitary gigantism, and Cushing syndrome can be part of the syndrome. Comments: The mosaic distribution of the GNAS1 mutation in different tissues results in the peculiar occurrence of skin, skeletal and endocrine abnormalities. In the ovary autonomous functioning cysts will initiate precious puberty. 30 Nevo syndrome Phenotype: Increased growth, kyphosis, prominent forehead, volar oedema, spindle-shaped fingers, wrist drop, talipes, hyperbilirubinemia, and generalised hypotonia. Developmental delay in some but not all cases reported. Long face, large extremities, cryptorchidism. Comment: Mutation in the gene encoding lysyl hydroxylase (PLOD1) and is therefore allelic with, or in the view of some identical to, the kyphoscoliotic type of Ehlers-Danlos syndrome. 31 Noonan syndrome Phenotype: Autosomal-dominant dysmorphic syndrome characterised by Turner-like dysmorphic features (chest deformity, a short neck with webbing or redundancy of skin, hypertelorism, epicanthic folds, ptosis, downslanting palpebral fissures, and low-set posteriorly rotated ears). Other features include short stature (50%), cardiac malformations (typically: pulmonary artery stenosis), deafness, motor delay, moderate mental retardation, behavioural problems, a bleeding diathesis, visual disturbance, hearing impairment, often cryptorchidism, anorchia, testicular atrophy, defective pubertal development, decreased fertility occurrence in males and females (1:1). In adulthood FSH (N, +), LH (N, +), T (N, –). Comments: One form of Noonan syndrome, that which maps to 12q24.1, is due to mutations in PTPN11, a gene encoding the nonreceptor protein tyrosine phosphatase SHP2, which contains 2 Src homology-2 (SH2) domains. Mutations in the PTPN11 gene account for about half the patients studied. Mutations in the neurofibromin gene (NF1), which is the site of mutations causing classic neurofibromatosis type I, have been found in neurofibromatosis-Noonan syndrome (NFNS). De novo germline mutations of the KRAS gene have also been found in individuals with Noonan syndrome (NS3), but account for less than 5% of Noonan syndrome cases. A form of Noonan syndrome (NS4) is caused by mutation in the SOS1 gene. Sporadic (80%) or autosomal dominant (20%). 1:1,000–2,500 livebirths. 32 Prader-Willi-Labhart syndrome or Prader-Willi syndrome (PWS) Synonyms: Hyperphagia, hypotonia, hypogonadism, obesity (HHHO) syndrome. Phenotype: (1) Diminished foetal activity, neonatal/infantile central hypotonia; (2) feeding problems/failure to thrive in infancy; (3) weight gain and hyperphagia after 12 months, obesity; (4) narrow bifrontal diameter, almond-shaped eyes; (5) moderate developmental delay (IQ <80); (6) hypogonadism, incomplete pubertal development; (7) short stature in approximately 50% of cases; (8) small hands and feet. Hypogonadotrophic hypogonadism: FSH (N, +, –), LH (N, +, –), testosterone (N, –). Insulin resistance due to obesity. Comments: Autosomal-dominant disorder caused by deletion or disruption of a gene or several genes on the proximal long arm of the paternal chromosome 15 or maternal uniparental disomy 15, because the gene(s) on the maternal chromosome(s) 15 are virtually inactive through imprinting. ESPE Classification of Paediatric Endocrine Diagnoses 114 Contiguous gene syndrome resulting from deletion of the paternal copies of the imprinted SNRPN gene, the necdin gene, and possibly other genes. 33 Proteus syndrome Phenotype: Hemihypertrophy/partial gigantism particularly affecting hands and feet; exostoses of skull, ear canals, nasal bridge, alveolar ridge; macrocephaly, skull asymmetry; vascular abnormalities (verrucous hamartomas, haemangiomas, lymphangiomas). Developmental delay in 50%. The skin changes are papillomatous epidermal nevi. Comments: Somatic mosaicism with alterations in genes affecting muscle differentiation/paracrine growth factors. Imaging studies useful in distinguishing from hemihyperplasia. A germline mutation in the tumour suppressor gene PTEN was found in 1 patient with Proteus syndrome. 34 Von Recklinghausen’s disease (type 1), neurofibromatosis type 1 (NF1) Synonyms: Neurofibromatosis, NF1 microdeletion syndrome. Phenotype: Neurofibromatosis is an autosomal-dominant disorder characterised particularly by cafe-au-lait spots and fibromatous tumours of the skin. Other features are variably present. Criteria: (1) six or more café-au-lait spots >0.5 cm (prepubertal) or >1.5 cm (pubertal) in diameter; (2) two or more neurofibromas (any), or at least one plexiforme neurofibroma; (3) ‘freckling’ in axilla; (4) glioma nervi optici; (5) bone lesions; (6) one 1st-degree relative with NF1. Benign tumours of perineural tissue (Schwann cells and other cells). Comments: Neurofibromatosis type I (NF1) is caused by mutation in the neurofibromin gene. Several young children with some features of NF1 and hematologic malignancies have been identified with homozygous mutations in the mismatch repair genes MLH1 and MSH2. Autosomal dominant, 50% new mutations. Incidence approximately 1:2,500. 35 Rieger syndrome Phenotype: Hypodontia (partial anodontia), malformation of the anterior chamber of the eye (microcornea with opacity, hypoplasia of the iris, and anterior synechiae), myotonic dystrophy, anal stenosis, characteristic facies. It has been suggested to label the combination of short stature, facial anomalies, Rieger anomaly, midline anomalies, and enamel defects the short-FRAME syndrome. Comments: There may be more than one genetic form of Rieger syndrome. Rieger syndrome type 1 is caused by mutations in a homeobox transcription factor gene, PITX2. Linkage studies indicated that a second type of Rieger syndrome maps to chromosome 13q14 (RIEG2). Autosomal dominant inheritance. 36 Rubinstein-Taybi syndrome Phenotype: Mental retardation, broad halluces, broad thumbs, facial abnormalities, congenital or juvenile glaucoma, persistent foetal pads of the fingers, shawl scrotum, frequent fractures, constipation, difficulties in sleep and anaesthesia by easily collapsible laryngeal walls, short upper lip, pouting lower lip, high slit-like palate. Comment: Can be caused by mutation in the gene encoding the transcriptional coactivator CREBbinding protein (CREBBP) or in the EP300 gene. Syndromes with Endocrine Features 115 37 Septo-optic dysplasia (De Morsier syndrome) Phenotype: Growth retardation, visual impairment, nystagmus; hypothalamic dysfunction and pituitary failure may occur. Neonatal hypoglycaemia and seizures. Developmental anomalies of the midline structures of the brain like hypoplasia of optic nerves, agenesis of septum pellucidum and agenesis of corpus callosum. Variable pituitary hormone deficiencies. Very variable phenotype. Comments: HESX1 mutations were found only in a few cases. 38 Silver-Russell syndrome (SRS) or Russell-Silver syndrome Phenotype: Congenital hemihypertrophy, low birth weight, short stature (postnatal shortness with an adult height of about –4 SDS), and elevated urinary gonadotrophins, characteristic facial features, including triangular shaped face with a broad forehead and pointed, small chin with a wide, thin mouth; clinodactyly V; 4. asymmetry. Variable phenotype. Comment: Chromosomal analysis: sporadic, maternal uniparental disomy of chromosome 7 has been described in approximately 10% of the cases. 39 Simpson-Golabi-Behmel syndrome Phenotype: Pre- and postnatal overgrowth with mild-to-moderate psychomotor delay. Hypotonia with macrocephaly. Postaxial polydactyly, partial syndactyly. Macroglossia, cleft lip/palate, hypertelorism. Cardiac conduction defects. Gastro-intestinal defects. Ambiguous genitalia. Renal defects with enlarged cystic kidneys, dysplasia, duplex systems, hydronephrosis, increased risk of embryonal tumours. Lab: Increased maternal alphafoetoprotein. Disorder of glypican (GPC3 + 4) metabolism. Comment: Similar to Beckwith-Wiedemann syndrome but distinguished by facial dysmorphism, broad thumb and great toe, and polydactyly. 40 Smith-Lemli-Opitz syndrome Phenotype: Hypospadias, failure to thrive, mental retardation, photosensitivity, abnormal sleep pattern, microcephaly, short or proximally placed thumbs, syndactyly, second-third toe syndactyly, congenital cardiac abnormalities (e.g. atrioventricular septal defect), visceral abnormalities. The typical facial appearance (coarse facies) becomes less obvious with age. Variable phenotype of the external genitalia. Serum 7-dehydrocholesterol level does not correlate with clinical severity. Comments: Caused by mutations in the sterol delta-7-reductase gene (DHCR7). This syndrome is the first true metabolic syndrome of multiple congenital malformations. Frequency: Approximately 1 in 20,000 to 30,000 births in populations of northern and central European background. 41 Sotos syndrome (cerebral gigantism) Phenotype: Accelerated prenatal and infantile growth results in macrocephaly and rapid childhood growth with bone age advance. Adult height not excessive – mean 173 and 184 cm in females and males, respectively. Developmental delay. Poor co-ordination. Unusual facies with long face, prominent chin and prominent forehead. Comment: NSD1 haploinsuffiency in most characteristic cases. 42 Steinert’s disease, myotonic dystrophy Phenotype: Myotonia, muscular dystrophy, cataracts, hypogonadism, frontal balding, and ECG changes. Comment: Myotonic dystrophy-1 (DM1) is caused by mutation in the dystrophia myotonica protein kinase gene (DMPK). The genetic defect in DM1 results from an amplified trinucleotide repeat in the ESPE Classification of Paediatric Endocrine Diagnoses 116 3-prime untranslated region of the protein kinase gene. Disease severity varies with the number of repeats: normal individuals have 5–30 repeats, mildly affected persons have 50–80 repeats, and severely affected individuals have 2,000 or more copies. Amplification is frequently observed after parent-to-child transmission, but extreme amplifications are not transmitted through the male line. This mechanism explains genetic anticipation and the occurrence of the severe congenital form almost exclusively in the offspring of affected women. Dystrophia myotonica-2 (DM2l) is caused by mutation in the ZNF9 gene. 43 Weaver syndrome Phenotype: Primordial overgrowth syndrome with prenatal and infantile growth acceleration; hypertonia and developmental delay. Camptodactyly, with thin, deep-set nails and prominent finger tip pads. Craniofacial dysmorphism (round face, hypertelorism, long philtrum, micrognathia, large ears, down-slanting palpebral fissures). Talipes equinovarus. Radiology: Accelerated skeletal maturation, broad distal femur and ulna. Comment: Some patients have a mutation in the NSD1 gene. 44 Williams-Beuren syndrome Phenotype: Supravalvular aortic stenosis, multiple peripheral pulmonary arterial stenoses, elfin face, mental and statural deficiency, characteristic dental malformation, and infantile hypercalcaemia. Comment: The disorder is a contiguous gene syndrome, autosomal-dominant inheritance. GTF2IRD1 and GTF2I are responsible for the main aspects of WBS, but other genes may also be involved (LIMK1, RFC2, CYLN2). 45 Wolfram syndrome (DIDMOAD) Phenotype: (Partial) Diabetes Insipidus, gradual onset of Diabetes Mellitus, Optic Atrophy, Deafness. In addition, neurogenic bladder, ataxia, psychiatric disorders. Signs outside the nervous system: hypogonadism, pigmented retinopathy, cardiomyopathy, sideroblastic anaemia, thrombocytopenia. Frequently insulin dependence, no autoimmune phenomena. Serum AVP (–). DNA: heterogeneous with mitochondrial and nuclear mutations. Comments: Caused by mutation in the gene encoding wolframin (WFS1). Another locus for the disorder has been mapped to 4q (WFS2). 46 Glucocorticoid excess (Cushing syndrome) Phenotype: Truncal obesity and moon facies, acne, hypertension, striae, loss of muscle mass, decreased growth velocity, emotional instability. Cortisol (+), androgens (+), estrogens (N/+), aldosterone (+), ACTH (+/N/–). Elevated blood glucose, hyperlipidaemia, insulin-resistant diabetes. Comment: Symptoms of hypercortisolism are similar whatever its cause. 47 Mauriac syndrome Phenotype: Short stature and glycogen-loaded liver in poorly controlled diabetes mellitus. Syndromes with Endocrine Features 117 48 Polycystic ovary syndrome (PCO) Synonym: Stein-Loewenthal syndrome. Phenotype: Menstrual irregularity, amenorrhea, anovulatory cycles, hirsutism, obesity, decreased fertility. Enlarged ovaries with multiple cysts, hyperthecosis. LH (+), FSH (N), LH:FSH ratio >3, T (+), adrenal androgens (+), Prl (+), insulin resistance. Comments: Not observed in prepubertal girls. No clear-cut diagnostic criteria during puberty. 49 Autoimmune polyglandular syndrome type 1 Synonym: Autoimmune polyendocrinopathy syndrome (APS)1, autoimmune polyendocrine syndrome, type I, autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED), polyglandular autoimmune syndrome, type I (PGA I), hypoadrenocorticism with hypoparathyroidism and superficial moniliasis, polyglandular deficiency syndrome, autoimmune polyendocrinopathy syndrome, type I. Phenotype: 2 of 3 major clinical symptoms: Addison’s disease, and/or hypoparathyroidism, and/or chronic mucocutaneous candidiasis. Comment: Caused by mutation in the autoimmune regulator gene (AIRE). 50 Autoimmune polyglandular syndrome type 2 Phenotype: Defined by the presence of adrenalitis with either autoimmune thyroid disease (Schmidt syndrome) or type 1 diabetes mellitus. Various combinations of other features, such as gonadal atrophy, hypoparathyroidism, alopecia, myasthenia gravis, pernicious anaemia, vitiligo, and Graves’ disease. Comments: The syndromal association may be due to unusual susceptibility to immunologic derangement because of a particular immune-response gene linked to (i.e. in linkage disequilibrium with) HLA on chromosome 6. 51 Autoimmune polyglandular syndrome type 3 Comment: Defined by the association between autoimmune thyroid disease and autoimmune disorders other than Addison’s disease. 52 Immune dysregulation, polyendocrinopathy, and enteropathy (X-linked) syndrome (IPEX) Phenotype: X-linked syndrome with various combinations of intractable diarrhoea, eczema, haemolytic anaemia, diabetes mellitus, or thyroid autoimmunity. Exaggerated responses to viral infections have been noted. Comment: Caused by mutation in the FOXP3 gene. 53 Autoimmune lymphoproliferative syndrome (APLS) Phenotype: Lymphadenopathy and splenomegaly associated with autoimmune haemolytic anaemia and thrombocytopenia. Comments: Type IA is caused by mutations in the FAS gene (TNFRSF6, or CD95), and type IB by mutations in the FAS ligand (FASL) gene (TNFSF6 or CD95L). Type IIA ALPS (ALPS2A) is caused by mutations in the caspase-10 gene (CASP10), and type IIB by mutations in the CASP8 gene. Type III ALPS comprises cases in which a mutation has not been identified. ESPE Classification of Paediatric Endocrine Diagnoses 118 54 Kabuki make-up syndrome Phenotype: Congenital mental retardation syndrome with additional features, including postnatal dwarfism, a peculiar facies characterised by long palpebral fissures with eversion of the lateral third of the lower eyelids (reminiscent of the make-up of actors of Kabuki, a Japanese traditional theatrical form), a broad and depressed nasal tip, large prominent earlobes, a cleft or high-arched palate, scoliosis, short fifth finger, persistence of fingerpads, radiographic abnormalities of the vertebrae, hands, and hip joints, and recurrent otitis media in infancy. 55 MEN1 Synonyms: Multiple endocrine neoplasia, type Ia (MEN1a, MEN I); multiple endocrine adenomatosis (MEA I), Wermer syndrome. Phenotype: Autosomal-dominant disorder characterised by a high frequency of peptic ulcer disease and primary endocrine abnormalities involving the pituitary, parathyroid, and pancreas. Comments: MEN1A is caused by a mutation in the MEN1 gene. MEN1B is caused by a mutation in the CDKN1B gene. 56 MEN2A Phenotype: Multiple endocrine neoplasia, type IIA, is an autosomal-dominant syndrome of multiple endocrine neoplasms, including medullary thyroid carcinoma, phaeochromocytoma, and parathyroid adenomas. Comment: Caused by a mutation in the RET oncogene. 57 MEN2B Phenotype: Multiple true neuromas, phaeochromocytoma, thyroid carcinoma (medullary type), sometimes café-au-lait spots. The neuromas occur as pedunculated nodules on the eyelid margins, lips and tongue. The lips are diffusely hypertrophied. Features of Marfan syndrome (high arched palate, pectus excavatum, bilateral pes cavus, high patella and scoliosis), nodular goitre, pigmentation of hands, feet and circumoral area, proximal myopathy, loose motions, and flushing attacks. Comment: MEN2B is caused by a mutation in the RET gene. Syndromes with Endocrine Features 119