Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Genome (book) wikipedia , lookup
Epigenetics of human development wikipedia , lookup
Ridge (biology) wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Metagenomics wikipedia , lookup
Genome evolution wikipedia , lookup
Pathogenomics wikipedia , lookup
Minimal genome wikipedia , lookup
Microevolution wikipedia , lookup
Gene expression profiling wikipedia , lookup
Biology and consumer behaviour wikipedia , lookup
Gene expression programming wikipedia , lookup
Quantitative comparative linguistics wikipedia , lookup
V13 Prediction of Phylogenies based on single genes
Material of this lecture taken from
- chapter 6, DW Mount „Bioinformatics“
and from Julian Felsenstein‘s book.
A phylogenetic analysis of a family of related
nucleic acid or protein sequences is a determination
of how the family might have been derived during
evolution.
Placing the sequences as outer branches on a tree,
the evolutionary relationships among the sequences
are depicted.
Phylogenies, or evolutionary trees, are the basic structures to describe
differences between species, and to analyze them statistically.
They have been around for over 140 years.
Statistical, computational, and algorithmic work on them is ca. 40 years old.
13. Lecture WS 2004/05
Bioinformatics III
1
3 main approaches in single-gene phylogeny
- maximum parsimony
- distance
- maximum likelihood
Popular programs:
PHYLIP (phylogenetic inference package – J Felsenstein)
PAUP (phylogenetic analysis using parsimony – Sinauer Assoc
13. Lecture WS 2004/05
Bioinformatics III
2
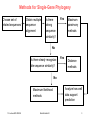
Methods for Single-Gene Phylogeny
Choose set of
related sequences
Obtain multiple
sequence
alignment
Is there
strong
sequence
similarity?
Yes
Maximum
parsimony
methods
No
Is there clearly recognizable sequence similarity?
Yes
Distance
methods
No
Maximum likelihood
methods
13. Lecture WS 2004/05
Bioinformatics III
Analyze how well
data support
prediction
3
Parsimony methods
Edwards & Cavalli-Sforza (1963): that evolutionary tree is to be preferred that
involves „the minimum net amount of evolution“.
seek that phylogeny on which, when we reconstruct the evolutionary events
leading to our data, there are as few events as possible.
(1) We must be able to make a reconstruction of events, involving as few events
as possible, for any proposed phylogeny.
(2) We must be able to search among all possible phylogenies for the one or
ones that minimize the number of events.
13. Lecture WS 2004/05
Bioinformatics III
4
A simple example
Suppose that we have 5 species,
each of which has been scored for 6 characters (0,1)
We will allow changes 0 1 and 1 0.
The initial state at the root of a tree may be either state 0 or state 1.
13. Lecture WS 2004/05
Bioinformatics III
5
Evaluating a particular tree
To find the most parsimonious tree, we must have a way of calculating how many
changes of state are needed on a given tree.
This tree represents the phylogeny of character 1.
Reconstruct phylogeny of character 1 on this tree.
13. Lecture WS 2004/05
Bioinformatics III
6
Evaluating a particular tree
There are 2 equally good reconstructions,
each involving just one change of character state.
They differ in which state they assume at the root of the tree,
and they differ in which branch they place the single change.
13. Lecture WS 2004/05
Bioinformatics III
7
Evaluating a particular tree
3 equally good reconstructions for character 2, which needs two changes of state.
13. Lecture WS 2004/05
Bioinformatics III
8
Evaluating a particular tree
A single reconstruction for character 3, involving one change of state.
13. Lecture WS 2004/05
Bioinformatics III
9
Evaluating a particular tree
on the right: 2 reconstructions for character 4 and 5 because these characters have
identical patterns.
single reconstruction for character 6,
one change of state.
13. Lecture WS 2004/05
Bioinformatics III
10
Evaluating a particular tree
The total number of changes of character state needed on this tree is
1+2+1+2+2+1=9
Reconstruction of the changes in state on this tree
13. Lecture WS 2004/05
Bioinformatics III
11
Evaluating a particular tree
Alternative tree with only 8 changes of state.
The minimum number of changes of state would be 6, as there are 6 characters that
can each have 2 states.
Thus, we have two „extra“ changes called „homoplasmy“.
13. Lecture WS 2004/05
Bioinformatics III
12
Evaluating a particular tree
Figure right shows another tree also requiring 8 changes. These two most
parsimonious trees are the same tree when the roots of the tree are removed.
13. Lecture WS 2004/05
Bioinformatics III
13
Methods of rooting the tree
There are many rooted trees, one for each branch of this unrooted tree,
and all have the same number of changes of state.
The number of changes of state only depends on the unrooted tree, and not at all on
where the tree is then rooted.
Biologists want to think of trees as rooted
need method to place the root in an otherwise unrooted tree.
(1) Outgroup criterion
(2) Use a molecular clock.
13. Lecture WS 2004/05
Bioinformatics III
14
Outgroup criterion
Assumes that we know the answer in advance.
Suppose that we have a number of great apes,
plus a single old-world monkey.
Suppose that we know that the great apes are a monophyletic group.
If we infer a tree of these species, we know that the root must be placed on the
lineage that connects the old-world monkey (outgroup) to the great apes (ingroup).
13. Lecture WS 2004/05
Bioinformatics III
15
Molecular clock
If an equal amount of changes were observed on all lineages, there should be a
point on the tree that has equal amounts of change (branch lengths) from there to
all tips.
With a molecular clock, it is only the expected amounts of change that are equal.
The observed amounts may not be.
using various methods find a root that makes the amounts of change
approximately equal on all lineages.
13. Lecture WS 2004/05
Bioinformatics III
16
Branch lengths
Having found an unrooted tree, locate the changes on it and find out how many
occur in each of the branches.
The location of the changes can be ambiguous.
average over all possible reconstructions of each character for which there is
ambiguity in the unrooted tree.
Fractional numbers in some branches of left tree
add up to (integer) number of changes (right)
13. Lecture WS 2004/05
Bioinformatics III
17
Open questions
* Particularly for larger data sets, need to know how to count number of changes
of state by use of an algorithm.
* need to know algorithm for reconstructing states at interior nodes of the tree.
* need to know how to search among all possible trees for the most parsimonious
ones, and how to infer branch lengths.
* sofar only considered simple model of 0/1 characters.
DNA sequences have 4 states, protein sequences 20 states.
* Justification: is it reasonable to use the parsimony criterion?
If so, what does it implicitly assume about the biology?
* What is the statistical status of finding the most parsimonious tree?
Can we make statements how well-supported it is compared to other trees?
13. Lecture WS 2004/05
Bioinformatics III
18
Counting evolutionary changes
2 related dynamic programming algorithms: Fitch (1971) and Sankoff (1975)
- evaluate a phylogeny character by character
- for each character, consider it as rooted tree, placing the root wherever seems
appropriate.
- update some information down a tree; when we reach the bottom, the number of
changes of state is available.
Do not actually locate changes or reconstruct interior states at the nodes of the tree.
13. Lecture WS 2004/05
Bioinformatics III
19
Fitch algorithm
intended to count the number of changes in a bifurcating tree with nucleotide
sequence data, in which any one of the 4 bases (A, C, G, T) can change to any
other.
At the particular site, we have observed the bases C, A, C, A and G in the 5 species.
Give them in the order in which they appear in the tree, left to right.
13. Lecture WS 2004/05
Bioinformatics III
20
Fitch algorithm
For the left two, at the node that is their immediate common ancestor,
attempt to construct the intersection of the two sets.
But as {C} {A} = instead construct
the union {C} {A} = {AC} and count 1
change of state.
For the rightmost pair of species, assign
common ancestor as {AG},
since {A} {G} = and count another
change of state.
.... proceed to bottom
Total number of changes = 3. Algorithm works on arbitrarily large trees.
13. Lecture WS 2004/05
Bioinformatics III
21
Complexity of Fitch algorithm
Fitch algorithm can be carried out in a number of operations that is proportional to
the number of species (tips) on the tree.
Don‘t we need to multiply this by the number of sites n ?
Any site that is invariant (which has the same base in all species, e.g. AAAAA) can
be dropped.
Other sites with a single variant base (e.g. ATAAA) will only require a single change
of state on all trees. These too can be dropped.
For sites with the same pattern (e.g. CACAG) that we have already seen, simply
use number of changes previously computed.
Pattern following same symmetry (e.g. TCTCA = CACAG) need same number of
changes numerical effort rises slower than linearly with the number of sites.
13. Lecture WS 2004/05
Bioinformatics III
22
Sankoff algorithm
Fitch algorithm is very effective – but we can‘t understand why it works.
Sankoff algorithm: more complex, but its structure is more apparent.
Assume that we have a table of the cost of changes cij between each character
state i and each other state j.
Compute the total cost of the most parsimonious combinations of events by
computing it for each character.
For a given character, compute for each node k in the tree a quantity Sk(i).
This is interpreted as the minimal cost, given that node k is assigned state i,
of all the events upwards from node k in the tree.
13. Lecture WS 2004/05
Bioinformatics III
23
Sankoff algorithm
If we can compute these values for all nodes,
we can also compute them for the bottom node in the tree.
Simply choose the minimum of these values
S min S 0 i
i
which is the desired total cost we seek, the minimum cost of evolution for this
character.
At the tips of the tree, the S(i) are easy to compute. The cost is 0 if the observed
state is state i, and infinite otherwise.
If we have observed an ambigous state, the cost is 0 for all states that it could be,
and infinite for the rest.
Now we just need an algorithm to calculate the S(i) for the immediate common
ancestor of two nodes.
13. Lecture WS 2004/05
Bioinformatics III
24
Sankoff algorithm
Suppose that the two descendant nodes are called l and r (for „left“ and „right“).
For their immediate common ancestor, node a, we compute
Sa i min cij Sl j min cik Sr k
j
k
The smallest possible cost given that node a is in state i is the cost cij of going from
state i to state j in the left descendant lineage, plus the cost Sl(j) of events further up
in the subtree gien that node l is in state j. Select value of j that minimizes that sum.
Same calculation for right descendant lineage sum of these two minima is the
smallest possible cost for the subtree above node a, given that node a is in state i.
Apply equation successively to each node in the tree, working downwards.
Finally compute all S0(i) and use previous eq. to find minimum cost for whole tree.
13. Lecture WS 2004/05
Bioinformatics III
25
Sankoff algorithm
The array (6,6,7,8) at the bottom of the tree has a minimum value of 6
= minimum total cost of the tree for this site.
13. Lecture WS 2004/05
Bioinformatics III
26
Finding the best tree by heuristic search
The obvious method for searching for the most parsimonious tree is to consider ALL
trees and evaluate each one.
Unfortunately, generally the number of possible trees is too large.
use heuristic search methods that attempt to find the best trees without looking at
all possible trees.
(1) Make an initial estimate of the tree and make small rearrangements of it
= find „neighboring“ trees.
(2) If any of these neighbors are better, consider them and continue search.
13. Lecture WS 2004/05
Bioinformatics III
27
Nearest-neighbor interchanges
13. Lecture WS 2004/05
Bioinformatics III
28
Nearest-neighbor interchanges
13. Lecture WS 2004/05
Bioinformatics III
29
Subtree pruning and regrafting
13. Lecture WS 2004/05
Bioinformatics III
30
Branch-and-Bound
find global optimum, NP-hard problem
13. Lecture WS 2004/05
Bioinformatics III
31
Resolve Incongruences in Phylogeny
Many possible reasons that may make decisions on how to handle conflicts in
larger sets of molecular data difficult.
E.g. two genes with different evolutionary history (e.g. owing to hybridization or
horizontal transfer) will necessarily give incongruent pictures while still depicting
true histories.
Here: compare genome sequence data for 7 Saccharomyces yeast species:
S. cerevisae
S. paradoxus
S. mikatae
S. kudriavzevii
S. bayanus
S. castelli
S. kluyveri
plus one outgroup fungus Candida albicans.
Rokas et al. Nature 425, 798 (2003)
13. Lecture WS 2004/05
Bioinformatics III
32
Resolve Incongruences in Phylogeny
Identify orthologous genes to serve as phylogenetic markers:
106 genes which are distributed throughout the S. cerevisae genome on all 16
chromosomes and comprise a total length of 127026 nt = 42342 amino acids
corresponding to roughly 1% of the genomic sequence and 2% of the predicted
genes.
Criteria to select genes spaced ca. every 40 kb:
(1) genes have homologous sequence in each of the 8 species
(2) genes have at least two homologous flanking syntenic genes
(3) genes can be aligned over most of the protein.
3 types of analysis:
- maximum likelihood (ML) analysis of nucleotide data
- maximum parsimony (MP) analysis of nucleotide data
- MP of the amino acid data
Rokas et al. Nature 425, 798 (2003)
13. Lecture WS 2004/05
Bioinformatics III
33
Resolve Incongruences in Phylogeny
Align individual genes with ClustalW. Edit manually to exclude indels and areas of
uncertain alignment left with 76% of the sequence of each gene on average.
Tree construction with PAUP by branch-and-bound algorithm which guarantees to
find the optimal tree. Estimate tree reliability using non-parametric bootstrap resampling.
Analysis of the 106 genes gave more than 20 alternative ML or MP trees.
Generate 50% majority-rule consensus trees by bootstrapping.
Next slide shows several strongly supported trees.
Rokas et al. Nature 425, 798 (2003)
13. Lecture WS 2004/05
Bioinformatics III
34
Bootstrap analysis.
A method for testing how well a particular data set fits a model.
E.g. the validity of the branch arrangement in a predicted phylogenetic tree can
be tested by resampling columns in a multiple sequence alignment to create
many new alignments.
The appearance of a particular branch in trees generated from these resampled
sequences can then be measured.
Alternatively, a sequence may be left out of an analysis to determine how
much the sequence influences the results of an analysis.
Here: swap individual nucleotide sites or positions of genes (bootstrap replicas).
13. Lecture WS 2004/05
Bioinformatics III
35
Alternative Tree topologies
Rokas et al. Nature 425, 798 (2003)
Single-gene data sets generate multiple, robustly supported alternative topologies.
Representative alternative trees recovered from analyses of nucleotide data of 106
selected single genes and six commonly used genes are shown. The trees are the
50% majority-rule consensus trees from the genes YBL091C (a), YDL031W (b),
YER005W (c), YGL001C (d), YNL155W (e) and YOL097C (f).
These 6 genes were selected without consideration of their function. Maybe
commonly used, well known genes of important functions provide a better resolution?
13. Lecture WS 2004/05
Bioinformatics III
36
Alternative Tree topologies
Results from the commonly used genes actin (g), hsp70 (h), -tubulin (i), RNA
polymerase II (j) elongation factor 1- (k) and 18S rDNA (l). Numbers above
branches indicate bootstrap values (ML on nucleotides/MP on nucleotides).
Same problem of alternative topologies as before.
Rokas et al. Nature 425, 798 (2003)
13. Lecture WS 2004/05
Bioinformatics III
37
Explanations?
The alternative phylogenies could have resulted from a number of different
scenarios:
(1) most genes could have weakly supported most phylogenies and strongly
supported only a few alternative trees,
(2) most genes could have strongly supported one phylogeny and a few genes
strongly supported only a small number of alternatives,
(3) there could have been some combinations of these scenarios so that each
branch among alternative phylogenies had either weak or strong support
depending on the gene.
To distinguish between these possibilities, identify all branches recovered during
single-gene analyses, record each bootstrap value with respect to the gene and
method of analysis.
8 branches were shared by all three analyses with multiple instances of
bootstrap values > 50%.
Rokas et al. Nature 425, 798 (2003)
13. Lecture WS 2004/05
Bioinformatics III
38
Common Branches
The distribution of bootstrap values for the eight prevalent branches recovered
from 106 single-gene analyses highlights the pervasive conflict among singlegene analyses. a, Majority-rule consensus tree of the 106 ML trees derived from
single-gene analyses. Across all analyses, there were eight commonly observed
branches; the five branches in the consensus tree (numbers 1–5; a) and the three
branches (numbers 6–8) shown in b.
Rokas et al. Nature 425, 798 (2003)
13. Lecture WS 2004/05
Bioinformatics III
39
Bootstrap Values of Common Branches
Only branches 1 and 4
are supported by a
majority of genes.
c, For each of the eight branches, the ranked distribution of per cent bootstrap values recovered from
the three analyses of 106 genes is shown. Results from ML (blue) and MP (red) analyses of
nucleotide data sets, and MP analyses of amino acid data sets (black), are shown. For each branch,
the mean bootstrap value and 95% confidence intervals from the ML analyses and the percentage of
ML trees supporting this branch (in parentheses) are indicated below each graph. Although the
ranked distributions of bootstrap values from the three analyses are remarkably similar for most
branches, on a gene-by-gene basis there is no tight correspondence between bootstrap values from
ML and MP analyses
Rokas et al. Nature 425, 798 (2003)
13. Lecture WS 2004/05
Bioinformatics III
40
How different are the trees?
The degree of conflict among the trees could be relatively minor.
Determine how many taxa (genes) would need to be removed to make two
trees congruent (deckungsgleich).
Rokas et al. Nature 425, 798 (2003)
13. Lecture WS 2004/05
Bioinformatics III
41
Reversal distance problem
Extensive incongruence between trees derived from
the 106 individual-gene data sets. Pairwise
comparisons between 50% majority-rule consensus
trees from 106 single-gene ML analyses of
nucleotide data (black bars), MP analyses of
nucleotide data (white bars), and MP analyses of
amino acid data (grey bars) were categorized on the
basis of the minimum number of taxa that need to be
removed for two trees to reach congruence (x axis).
For each of the analyses, the majority of
pairwise comparisons require the
removal of two or more taxa before
congruence is attained.
Rokas et al. Nature 425, 798 (2003)
13. Lecture WS 2004/05
Bioinformatics III
42
What leads to incongruence?
Many factors were checked that could lead to incongruence between single-gene
phylogenies:
- outgroup choice
repeat all analyses without C. albicans
- number of variable sites
significantly correlated with
- number of parsimony-informative sites
bootstrap values for some
- gene size
branches
- rate of evolution
- nucleotide composition
- base compositional bias
- genome location
- gene ontology
}
no parameters can systematically account for or predict the performance of single
genes!
Rokas et al. Nature 425, 798 (2003)
13. Lecture WS 2004/05
Bioinformatics III
43
Can incongruence be overcome?
Although we do not know the cause(s) of incongruence between single-gene
phylogenies, the critical question is how this incongruence between single trees
might be overcome to arrive at the actual species tree.
Can single gene trees be concatenated into one large data set?
Rokas et al. Nature 425, 798 (2003)
13. Lecture WS 2004/05
Bioinformatics III
44
Concatenation of single genes gives a single tree!
Phylogenetic analyses of the
concatenated data set composed
of 106 genes yield maximum
support for a single tree,
irrespective of method and type of
character evaluated. Numbers
above branches indicate bootstrap
values (ML on nucleotides/MP on
nucleotides/MP on amino acids).
All alternative topologies were rejected.
This level of support for a single tree with 5 internal branches is unprecedented.
This tree can now be referred to as species tree.
Rokas et al. Nature 425, 798 (2003)
13. Lecture WS 2004/05
Bioinformatics III
45
How much data is required?
The concatanated data recovered a tree with maximum support on all branches,
despite divergent levels of support for each branch among single-gene analyses.
At what size did the data set arrive at the species tree?
Rokas et al. Nature 425, 798 (2003)
13. Lecture WS 2004/05
Bioinformatics III
46
Convergence on single tree
branch 3
branch 5
A minimum of 20 genes is required to recover >95% bootstrap values for each
branch of the species tree. a, b, The bootstrap values for branches 3 (a) and 5 (b)
were constructed from the concatenation of randomly re-sampled orthologous
nucleotides (left) or random subsets of genes (right).
The species tree is recovered with robust support (>95% bootstrap values in all
branches at 95% confidence interval) by analyses of a minimum of 20
concatenated genes. All analyses were performed using MP.
Rokas et al. Nature 425, 798 (2003)
13. Lecture WS 2004/05
Bioinformatics III
47
Independent evolution?
It has been suggested that nucleotides within a given gene do not evolve
independently.
Re-sample subset of orthologous nucleotides from the total data set.
Only 3000 randomly chosen nucleotide positions (corresponding to less than three
concatenated genes) are sufficient to generate single tree with > 95% confidence.
This indicates that nucleotides in genes have not evolved independently (because
when using complete genes more than 20 genes are necessary to generate single
tree).
Rokas et al. Nature 425, 798 (2003)
13. Lecture WS 2004/05
Bioinformatics III
48
Implications for resolution of phylogenies
Unreliability of single-gene data sets stems from the fact that each gene is shaped
by a unique set of functional constraints through evolution.
Phylogenetic algorithms are sensitive to such constraints.
Such problems can be avoided with genome-wide sampling of independently
evolving genes.
In other cases the amount of sequence information needed to resolve specific
relationships will be dependent on the particular phylogenetic history under
examination.
Branches depicting speciation events separated by long time intervals may be
resolved with a smaller amount of data, and those depicting speciation events
separated by shorter invtervals may be much harder to resolve.
Rokas et al. Nature 425, 798 (2003)
13. Lecture WS 2004/05
Bioinformatics III
49
Summary
Robust strategies exist for phylogenies built on single-gene comparisons
(maximum parsimony, distance, maximum likelihood).
Problem of incongruence of phylogenies derived from individual genes.
Can be resolved by integrative analysis of multiple (here > 20) genes.
It is desirable to combine results from phylogenies constructed from local
sequence information with trees constructed from genome rearrangement.
The power of genome rearrangement studies is the construction of ancestral
genomes. Then one can derive the speed of evolution at different times, disect
mutation biases at different times from the influence of genomic context ...
and possibly derive the driving forces of biological evolution.
13. Lecture WS 2004/05
Bioinformatics III
50