Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
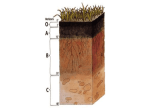
Abstract Dead plant biomass is a key pool of carbon in terrestrial ecosystems. Its decomposition in soil environments is thus an essential process of the carbon cycle. Fungi are considered to be the primary decomposers in soil ecosystems because of their physiological adaptations and enzymatic apparatus composed from highly effective oxidative and hydrolytic enzymes. Many recent works show that in addition to fungi, bacteria may also play a significant role in lignocellulose decomposition and among bacteria, the members of the phylum Actinobacteria are often regarded to significantly contribute to cellulose and lignocellulose decomposition. This thesis is focused on the evaluation of the role that fungi and Actinobacteria play in dead plant biomass degradation. First, it explored mechanisms involved in degradation, in particular the enzymatic breakdown of major lignocellulose components as cellulose, hemicelluloses and lignin. Enzymatic apparatus of the saprotrophic fungus Fomes fomentarius was explored both in vitro as well as in vivo. Several Actinobacteria were isolated from soil and comparative experiments, investigating production of hydrolytic enzymes, were carried out to track the transformation of polysaccharides and lignin by these strains. To explain the roles of lignocellulose decomposers in complex environments like soil, the community composition of fungi and bacteria, with special focus on Actinobacteria, was investigated. The use of next generation sequencing (NGS) methods to cover this task required implementation and design of appropriate tools for data handling, not available at the time when the studies were conducted. This resulted in the development of the software pipeline SEED. With the help of this tool, active degraders in forest soil were identified by the comparison of RNA and DNA communities. Further, it was demonstrated that some microbial taxa show high RNA/DNA ratio or were detected only in RNA pool and thus they are underestimated or missing in studies based on DNA analysis. Results also confirmed the importance of Actinobacteria showing that they belong to the most active bacterial groups especially in soil organic horizon. To provide an in-depth analysis of actinobacterial communities along the gradient of heavy metal contamination, where they were expected to represent the most metabolically active group, the method for a selective community composition analysis of the Actinobacteria using 454 pyrosequencing was developed that allows community composition at a high resolution. The study showed that diversity of Actinobacteria was unaffected by heavy metal content, but the contamination changed the community composition significantly. Results also confirmed that Actinobacteria thrive better than other bacteria in contaminated soils and may thus serve as important degraders of lignocellulose. Finally, to get the most accurate estimates of the relative abundance of fungal and bacterial taxa obtained by NGS, methods improving these estimates were proposed. This part of study investigated the impact of the variability of 16S copy numbers among the bacterial genomes on the diversity and abundances of different taxonomical groups and steps to refine the abundance estimates were suggested. In the case of fungal communities, an alternative marker derived from the single-copy gene rpb2 was compared with the widely used multicopy ITS with the aim to reduce the problems of community analysis due to the intragenome variability and multicopy nature of the latter marker. Broad taxonomic coverage, suitability for taxonomic assignments and sufficient variation for the use in phylogenetic analyses were confirmed for the newly proposed marker.