Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Cre-Lox recombination wikipedia , lookup
Molecular evolution wikipedia , lookup
Molecular Inversion Probe wikipedia , lookup
Metalloprotein wikipedia , lookup
SNP genotyping wikipedia , lookup
Deoxyribozyme wikipedia , lookup
Nucleic acid analogue wikipedia , lookup
Molecular ecology wikipedia , lookup
Community fingerprinting wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
DNA sequencing wikipedia , lookup
Whole genome sequencing wikipedia , lookup
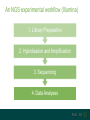
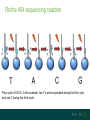
A brief introduction to... NGS and file formats Dr Laura Emery Overview • Terminology • Sequencing technologies • File formats NGS Terminology Many NGS technologies produce ‘short reads’ Sanger NGS genomic DNA DNA fragments long reads ~800bp short reads ~50-300bp Some terminology… • Depth: For any given base, the number of reads aligned at that positon • Coverage: the proportion of your target genome that is covered by some mapped sequence. Calculated as the number of reads * read length / assembly size E.g, 20x coverage means each base has been sequenced an average of 20 times More terminology… • Data volume – the data (in Gb) that can be obtained from a flow cell in one run of the machine. • Multiple samples can be loaded into different lanes within a flow cell • Run – The sequencing that you can do with the machine in one cycle (turning it on and off once) NGS Technologies NGS Technologies • A variety of platforms available • Differ in: • Library preparation • Sequencing chemistry Comparison of 2nd Gen NGS Technologies Library preparation Sequencing chemistry Features Roche 454 Emulsion PCR Pyrosequencing Longer read length, only available until 2016 Illumina HiSeq Solid phase amplification Reversible terminator Best output to cost ratio, low error rates Applied Biosciences SOLiD Emulsion PCR Sequencing by ligation Highest accuracy, very short reads Ion Torrent Solid phase amplification Proton detection Short run time, homopolymer errors An NGS experimental workflow (Illumina) 1. Library Preparation 2. Hybridisation and Amplification 3. Sequencing 4. Data Analyses 1. Illumina library preparation 1. Fragmentation and size selection 2. Adaptor ligation genomic DNA 2. Hybridisation and Amplification 3. Illumina sequencing reaction • DNA fragments attached to slide via adapters and amplified (PCR) prior to sequencing reaction • Nucleotides modified • Reservible terminators • Flourophore • Incoporated one at a time • Image taken, and terminators cleaved 3. Illumina sequencing reaction cycle 1 cycle 2 cycle 3, etc 3. Illumina sequencing reaction Roche 454 sequencing reaction • DNA fragments attached to beads via adapters and amplified (PCR) prior to sequencing reaction • Each bead in single well • One of 4 nucleotides added to well • Contain flourophore • Multiple nucleotides may be incorporated in each cycle • Image taken, and strength of signal reflects numbers of incorporated nucleotides Roche 454 sequencing reaction Flow cycle is TACG. In this example, two T’s are incorporated during the first cycle, and one C during the third cycle. Roche 454 signal output • Where 2 identical nucleotides have been incorporated, the detected signal is ~twice as strong • With longer homopolymer stretches, the determination of the exact number of identical bases becomes more error prone. TTCACTCGAACT Ion Torrent sequencing reaction Nucleotide incorporation measured by a change in pH from H+ ion release during reaction Third generation sequencing technologies • PacBio • single molecule sequencing, long reads, high error rate • Applications include whole genome shotgun, amplicon and transcriptomics • Oxford Nanopore • single molecule sequencing • long reads, high error rate • > Neither currently used in mainstream metagenomics studies due to high error rates PacBio vs Oxford Nanopore PacBio sequencing reaction Oxford Nanopore • Nanopore sensing based on disruption of ion flow through nanopore Oxford Nanopore Oford Nanopore mismatches Factors to consider when selecting a sequencing technology • Read length: longer better • More specific, less ambigious • More accurate alignments, classifications • Produce longer contigs when assembling • Accuracy: higher better (especially relevant for amplicon) • Data volume: higher better • More reads reduces the impact of errors • Necessary for WGS • Cost: lower better For a comparison, see Travis Glenn (2011) Field guide to next-generation DNA sequencers. Mol. Ecology Resources 11, 759-769 Summary of Platforms Sequencer Chemistry Read Length Volume Run Time Advantage Disadvantage Metagenomics? 454 GS FLX+ (Roche) Pyro- mode 700 up to 1 kb 700 Mb 23 hours Long reads High error rate Maybe HiSeq (Illumina) Reversible terminator 36 – 150 bp 95-600 40 hrs – days HighLow cost Short reads Long run time Yes MiSeq (Illumina) Reversible terminator 25 – 300 bp 13-15 5 hrs Short run time Short reads No SOLiD (Life) Ligation 50 – 75 bp 120-160 8 days Low error rate Short reads Long run time No Ion Torrent/ Proton (Life) Proton <200 bp 10 Gb 2 – 4 hr Short run time Chimeras, homopolymer Yes Oxford Ion flow disruption 5.4kb 150MB 6 hrs; fixed No PCR Very long reads Short run time High error rate No* PacBio RS Real time sequencing mean 4.6 kb to 23 kb 3GB 30 – min No PCR Very long reads Short run time High error rate No Summary of Platforms Sequencer Chemistry Read Length Volume Run Time Advantage Disadvantage Metagenomics? 454 GS FLX+ (Roche) Pyro- mode 700 up to 1 kb 700 Mb 23 hours Long reads High error rate Maybe HiSeq (Illumina) Reversible terminator 36 – 150 bp 95-600 40 hrs – days HighLow cost Short reads Long run time Yes SOLiD (Life) Ligation 50 – 75 bp 120-160 8 days Low error rate Short reads Long run time No Ion Torrent/ Proton (Life) Proton <200 bp 10 Gb 2 – 4 hr Short run time Chimeras, homopolymer Yes Oxford Ion flow disruption 5.4kb 150MB 6 hrs; fixed No PCR Very long reads Short run time High error rate No* PacBio RS Real time sequencing mean 4.6 kb to 23 kb 3GB 30 – min No PCR Very long reads High error rate No NGS File Formats NGS file formats from the machine Most common file formats: most widely used • FastQ (Illumina and others) • FastA (no quality information) • SFF (Standard Flowgram File) (Roche 454) Less common: • SRF (Helicos) • HDF5 (PacBio, Applied Biosystems, Oxford Nanopore) Note: NCBI SRA produces .sra files - use NCBI toolkit to convert . FastQ format FastQ tools • Many NGS tools handle FastQ • biopython: useful for creating separate fastA and quality score files (linux, mac OS X, windows) • FastQC: quality assessment of sequence runs (linux, mac OS X, windows) http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ • FASTX-Toolkit: collection of tools for FastA/FastQ preprocessing (linux, mac OS X) http://hannonlab.cshl.edu/fastx_toolkit/ • fastq-dump to extract fastq files from NCBI .sra files (linux, mac OS X, windows) http://www.ncbi.nlm.nih.gov/IEB/ToolBox/CPP_DOC/ What are Phred quality (Q) scores? • Quality scores show the probability of an erroneous base call • quality score Q = –10 log10 P • error probability P = 10–Q/10 • example: call with Q = 30 has error probability P = 10-3 = 1 in 1000 • Score range is from 0 – 93 (represented by ASCII code 33 – 126. • e.g. Q score ‘0’ can be represented as ‘!’, ’10’ as ‘+’ FastA Like FastQ, but with no quality information or + line: indicates new sequence starts, like @ of fastQ sequence name >ERR010482.1 FT9FZH301B6YPS/3 ATCAACACATTAGGACTTACACGAATCAGGCATTCGTTACCATCA sequence GTATGTCGAT >ERR010482.2 FT9FZH301ARSRC/3 ATGCTTGCTCGGCCGACGTGAGCGTTATTCGAGCAGGGCTCGGAT GGTAGTTAGCGATCCAAAGGGGAGTC SFF format (Roche 454) • Binary format • Contains (flow gram) signal strengths, bases and quality values • Contains information about the first and last base to be included after clipping adapters, barcodes (MIDs) and low-quality base calls Flowgram: 1.03 0.00 1.01 0.02 0.00 0.9 1.04 0.00 0.00 0.97 0.00 0.96 0.02 0.0 0.00 2.84 0.06 1.00 0.01 0.13 1.01 0.0 1.01 0.06 0.00 1.04 3.72 0.03... Flow Indexes: 1 3 6 8 10 13 15 18 20 2 31 34 35 37 37... Bases: tcagatcagacacgCCACTTTGCTCCCATTTCAGCACC CAACAAGAGCTTCCAAAGTTTCCCACCGGATCGACGGT TCTCCTTATCCTCAGCAGACAGCTTTGATGGACACGCT CAAAAGGATCACGATGATTCAACATGGCGCCAAACCAA Quality Scores: 40 40 40 40 40 40 40 4 40 40 40 40 40 38 38 38 40 40... • An detailed description can be found here: http://tinyurl.com/qcch8p4 How to interpret flowgram values (SFF) The start of the example flowgram has these signals: 1.03 0.00 1.01 0.02 0.00 0.96 0.00 1.00 0.00 1.04 0.00 0.00 0.97 0.00 0.96 0.02 0.00 1.04 0.01 1.04 0.00 0.97 0.96 0.02 0.00 1.00 0.95 1.04 0.00 0.00 2.04 0.02 0.03 1.05 0.99 0.01 After rounding: 1 0 1 0 0 1 0 1 0 1 0 0 1 0 1 0 0 1 0 1 0 1 1 0 0 1 1 1 0 0 2 0 0 1 1 0 With the flow order T|A|C|G, this translates into: 1 T's; 0 A's; 1 C's; 0 G's 0 T's; 1 A's etc, or TCAGATCAGACACGCCACTTT SFF format (Roche 454) tools available Some useful tools for SFF files: • Newbler tools (De Novo Assembler) on linux, free download from Roche http://www.454.com/products/analysis-software/ • • • • SFF Workbench (windows) http://www.dnabaser.com/download/SFF%20tools/ biopython (linux, mac OS X, windows) http://biopython.org/wiki/Main_Page sff2fastq (linux, mac OS X) https://github.com/indraniel/sff2fastq sff-dump to extract sff files from NCBI 454 .sra files (linux, mac OS X, windows) http://www.ncbi.nlm.nih.gov/IEB/ToolBox/CPP_DOC/ Many thanks to… The EBI training team • Paul Greenfield (CSIRO) • Peter Sterk • Mar Gonzàlez-Porta Thank you Images from EBI online training course “What is Next-Generation DNA Sequencing”