Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
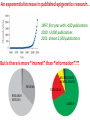
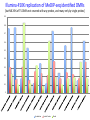
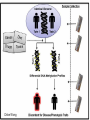
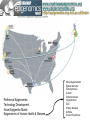
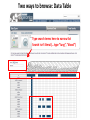
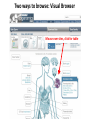
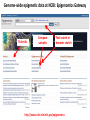
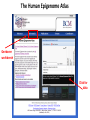
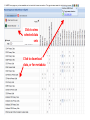
ERROR, BIAS, PROBLEMS AND PITFALLS IN EPIGENETIC EPIDEMIOLOGY DR JONATHAN MILL PSYCHIATRIC EPIGENETICS GROUP MRC SGDP CENTRE INSTITUTE OF PSYCHIATRY KING’S COLLEGE LONDON [email protected] www.epigenomicslab.com Newcastle, 2012 An exponential increase in published epigenetics research… 1997: first year with >100 publications 2010: >2,000 publications 2011: almost 2,500 publications But is there is more “interest” than “information”??? NEUROSCIENCE MENTAL HEALTH REVIEWS RESEARCH ARTICLES STEM CELLS CANCER “…The victory over the genes… Smarter, healthier, happier... How we can outwit our genome…” “…Roll over, Mendel. Watson and Crick? They are so your old man's version of DNA…” “…The integration of standard massage practices and the knowledge of the biology of adversity will change our minds, our physiology, our epigenetics—and hence our massage practice…” Would you trust these guys with your money? Behavioural epigenetics is highly controversial NATURE Vol 467|9 September 2010 AGTGCCTCAGCCTCCCTAGTAGCTGGGATTACAGGTGCCCTCCACAATGCCCAGCTAATTT TTGTGTTTTTAGTAGACACAAGATTTCACTATGTTGCCCAGGCTGGTCTCAACCCCTGACC TCAAGTGATCCACCTGCCTCAGTCTCCCAGAGTGCTGGGACTGCAGGCGTGAGCCAACAAG CCCAGGCCACGATGTCTTACTTTTCACCTAAAACCTGCCTAAATGGCATGCCCAGTTAAAA CAATCTTTTTCTGTTACAATAATCCATGTAAGAGTATGACACATTTTCTGAAAGATTTGTC TAAAAAAGAGCCTGGTATGTTTACTGTTGCTGCTGAATTGGATTTGACTCTGCTGCTGTAT CAGGGCCCCTTCTGACAATTCACCTCTTGCTTCCTTTCCTGCTAATTGTCCTGTTGACTAC – 1 body, 1 genome: a blood sample is all you need! TATTTTTTTTTTTTTTTGGTAACAGTGTCTGGCTCTGTCACCCAGCCTAGAGTGCAGTGGC – 1 life, 1 genome: you are born with the genome ACAATCTTGGCTCACTACAACCTCCATCTTCTGGGCTCAAGCTATTCTTCCACCTCAGCCT CCCAAGTAGCTGAGACTACAGGCATGTGCCACCACACCCAGCTAGATTTTGTATTTTTTGT you die with AGAGACGGGGTCTTGTGATGTTGCCCAGGCTGGTCTTGAACTCCTGGGCTCAAAGCAATCC – Any lifestyle, 1 genome: it doesn’t matter what GCCCGCCTCCGCCTCCCAAAGTGCTGAGATGACAGGCGTGAGCAACTGCGCCCAGCCTTGT GTACTTCTTAGGGCTCTTTTACATGCCTTTCTTTTTTTAACAGCCTTCCCACCACTACCTT you’re exposed to TTACATGTCTTGAGATTTTCCTGTATGCATGTGTATGCGTGCACGTGCACGCACGCACACA – Any disease, 1 genome: no reverse causation CACACACACACCTGATTTTGTCATTCTGGTGTTTAAAGCATATCATAGTCCTACTTCCAGA AATACATCCAATGCAATGAACCTGGTAGCCAACACTGCTGAGAAATGACCCAAGGGTCTAC – A nicely annotated reference genome and CTTGAGTAGCCAGCCCCCAAATCCAAAGAATAGCTCCAGACCCCATAGTTTTCTCACCCAC TAGGTCATGGGACCATGGCAAGAGTGAGAGAGTTCCACTTCCCAGAGGATGCCTGTTATTA catalogue of SNPs is freely available CCTTACCTCAATTTGAAATCTGTACTAAGGTTGAACACATGCATTCTCCTCCTTGACCTCC – Methods that do as they say on the box and give ACATCCCCTGTTGTTTCCTTTTTTTGTTGTTTTTGTTTTTTGTTTTTGTTTTGAGACAGAG TCTCGCTCTGTCGCCCAGGCTGGAGTGCAGTGGCACGATCTCGGCTCACTGCAGTCTCTGC results that are easy to interpret CTCCCGGGCTCAAGCAATTCTCCTGCCTCAGCCTCCTGAGTAACTGGGATTACAGGTGTGT Some of the (many) issues in epigenetic epidemiology • • • • • • • • • Technical / methodological Sample related issues Study design Analysis and interpretation Over-interpretation Over-simplification Biological Significance? Confounding factors Cause vs effect Schizophrenia Bipolar disorder Autism Spectrum Disorders Wong (in prep) Alzheimer’s Disease QDP ITFG2 Schalkwyk et al (in prep) How do different disease-relevant regions of the brain differ epigenetically? What is a ‘normal’ brain methylome? Are there marked epigenetic differences between the major cell-types in the brain? How is the brain methylome influenced by factors such as age, sex and medication? What is the location of functionallyrelevant DMRs? Can peripheral tissues be used as a ‘proxy’ for the brain in epigenetic epidemiology? IJE, 2012 Biological, Technological, and Methodological issues 1. We do not know where in the genome to look and what to look for 2. We have to rely on imperfect technology 3. We may be limited by available sample sizes that are optimal for epigenetic epidemiology 4. Whatever we do, it may never be enough to fully account for epigenetic differences between tissues and cells 5. We may be trying to detect inherently small effect sizes using sub-optimal methods and sample cohorts 6. We lack a framework for the analysis of genome-wide epigenetic data 7. We have to manage high expectations 1. We do not really know where to look, or what to look for Promoter CpG islands!!! CpG Island ‘Shores’ Irizarry et al, Nature Genetics, 2009 Most tissue-variable CGI DMRs: enrichment for intragenic CGIs 1000 900 Χ2 p = 1E-246 O/E=2.37 800 700 600 O/E=0.09 500 OBS EXP 400 300 200 100 0 Promoter Davies et al (in press) Intragenic 3'UTR Intergenic HCP Davies et al (in press) LCP And there’s more to epigenetic gene regulation than DNA methylation! Zhou et al, Nat Rev Genet, 2011 How many DNA modifications are there?? 5-hydroxymethylcytosine (5hmC) 5-formylcytosine (5fC) 5-carboxylcytosine (5caC) 5hmC appears to be particularly important in ES cell differentiation and the CNS and is implicated in postnatal neurodevelopment and aging Traditional bisulfite-based methods do not distinguish between 5-mC and 5hmC CMS (the product of bisulfite conversion of 5-hmC) tends to stall DNA polymerases during PCR densely hydroxymethylated regions of DNA may be underrepresented in quantitative methylation analyses Existing 5-mC data sets may require re-evaluation in the context of the possible presence of 5-hmC Antibodies against 5-mC and 5-hmC can pull out fragments enriched for each mark separately – but can’t quantify at base-pair resolution Oxidative bisulfite-sequencing (ox-BSseq) (Booth et al, Science, 2012) Huang et al, PLoS ONE, 2010 Lunnon et al (in prep) The relationship between DNA methylation and gene expression is not necessarily straightforward… (SAM) Beyond transcriptional silencing: functions of DNA methylation • Chromatin compaction • Genome stability • Suppression of homologous recombination between repeats • Genome defense against retroviruses • Genetic recombination & DNA mutability • X-chromosome inactivation (in females) • Genomic imprinting Gene-body DNA methylation Madeleine et al, Nature Biotechnology, 2009 41 (82%) out of the top 50 ranked cerebellum-cortex DMRs are mirrored by significant gene expression differences p = 1.90E-33 Cerebellum Frontal cortex DNA methylation Log2 expression EOMES p = 1.39E-34 Cerebellum Frontal cortex DNA methylation Log2 expression GRM4 2. We have to reply upon imperfect technology Measuring DNA methylation is not like measuring genotype Genome-scale assessment now feasible Compromise between coverage and precision Huge range of methods (enrichment, measurement and analysis) No consensus on analysis method There is a huge number of methylomic profiling methodologies Sensitivity Cost Reproducibility Coverage & Throughput Sample requirements Laird et al, 2010 Illumina 450K methylation array and EWAS Still very much focused on CpG Islands…. High correlation across genome-wide platforms….. But…mainly driven by the fact that the large majority of the genome is either unmethylated or fully methylated …substantial discrepancies between platforms may exist for intermediate level methylation Can these genome-based approaches detect a small % change? Many stages when inaccuracies in measuring DNA methylation may occur • Error (variation) can be introduced during – – – – – – – Tissue / cell processing DNA preparation / storage Enrichment / conversion Amplification Measurement QC Analysis • Accurate measurement may be vital if DNA methylation is to be used for clinical (i.e. diagnostic or prognostic) purposes • But is ‘accuracy’ vital for epidemiological studies? • Consistency / reliability more important? Some inconvenient truths • nothing you can do with normalization replaces careful experimental design • a nuisance variable confounded with what you want to test can’t be fixed – eg case and control in different batches • nothing you can do with normalization replaces rigorous QC – samples with unusual raw intensity distributions – multivariate methods such as PCA often identify mislabeled samples • Problems with outsourcing and core facilities • Bad data will always be bad data!! PCR bias: the effect of annealing temperature Make sure you PCR machines are calibrated!! PCR block Illumina batch effects Block 1 Block 2 Block 3 Block 4 The Illumina 450K array – some problems… • Normalisation issues – Type 1 and 2 probes – Batch effects – Array position • Colour collection • SNPs on probes • Cross-hybridising probes – Sex chromosomes very obvious 6-10% probes are non-specific SNPs in array probes common Features and sources of bias for DNA methylation technologies Laird, Nat Rev Genet, 2010 No method is perfect… • Bisulfite-based methods: PCR biases, bisulfite / PCR batch effects, hydroxymethylation • Affinity-based methods – CG density, IP efficiency / non-specificity, resolution, quantification, CNV confounds • MSRE-based methods – limited resolution, SNPs in MSRE sites • And it’s not all bad news especially for bigger effects. • For smaller differences – does non-verification negate a true difference? • Data integration and meta-analysis across studies MeDIP-seq vs 450K Illumina data Cerebellum Frontal cortex Whole blood 0.9 0.8 0.7 0.6 0.5 0.4 0.3 0.2 0.1 0 P = 6.73E-104 Cerebellum Frontal Cortex Blood Illumina 450K replication of MeDIP-seq identified DMRs [but NB 20% of TS-DMRs not covered with any probes, and many only by single probes] 0.9 0.8 0.7 0.6 0.5 0.4 0.3 0.2 0.1 0 Cerebellum Frontal Cortex Blood MeDIP-seq vs bisulfite pyrosequencing data • Low-frequency DNA methylation states may not be optimally detected by genomewide (sequencing-based) approaches because sensitivity is a function of readdepth • They may also not be detected via sitespecific approaches such as Pyrosequencing or Sequenom EpiTYPER (sensitivity 2-5%) • Ultra-deep bisulfite-sequencing of targeted regions? If something seems too good to be true….it probably is Things to look for when interpreting published data… • What enrichment/discrimination method has been used? • Have the relevant controls been used? • Have reactions been done in replicate (especially sodium bisulfite conversion, bisulfite PCR) • What magnitude of difference is being reported? • Do the differences reported exceed the sensitivity of the platform? F2RL3 encodes a protein that has functions which are relevant to cardiovascular disease Implications for confounding effects in epigenetic epidemiological analyses Sample-related technical issues… • • • • • Limited number of samples – overlap between studies? Pre-mortem factors and pH Cause of death Peri-mortem factors Post-mortem factors After a typical group lunch in the Mill lab… DNA preparation (phenol/chloroform, columns, etc) DNA storage (TE, Te, water, -80, -20, 4) Importance to keep consistent across samples (confounding case-control differences, longitudinal changes, etc) 26,320 years ago…. “…Our results suggest that as long as ancient nuclear DNA remains amplifiable, cytosine methylation patterns can be assessed…methylation has been faithfully retained along with the DNA over evolutionary timescales…” Llamas et al, PLoS ONE, 2012 3. We may be limited by available sample sizes that are optimal for epigenetic epidemiology The simple brute-force approach that has been used (relatively) successfully in GWAS is not valid for EWAS Simply running Illumina 450K arrays on your GWAS samples is potentially a waste of ££ Rakyan et al, 2011 Discordant Monozygotic Twins – a powerful tool for epigenetic studies of complex disease Control for: age, sex, genetics, pre-/perinatal environment, parental origin Verification of array data Replication in brain tissue Significant hypomethylation (20-30%) in 15% of SZ brains Dempster et al (2011) Chloe Wong Virtually Identical Large Changes DRD4 SERT Discordance for disease phenotypes?? MAOA Wong et al 2010 Longitudinal sampling… Most existing cohorts were not designed with epigenetics in mind Do sequential samples (of relevant tissues/cells) exist in ongoing longitudinal cohorts / biobanks? DNA methylation at three CpG sites—in the promoters of the EDARADD, TOM1L1 and NPTX2 genes—is linear with age over a range of five decades. Regression model that explains 73% of the variance in age, and is able to predict the age of an individual with an average accuracy of 5.2 years! Age is a huge potential confounder in epigenetic studies Ruth Pidsley (unpublished) Bell et al, PLoS Genetics, 2012 Slide No. 62 Boks et al (Epigenetics, in press) CRESTAR Kick-off Meeting 2011 4. Whatever we do, it may never be enough to fully account for epigenetic differences between tissues and cells Davies et al, in press Machine learning 100% tissue discrimination HOXA gene cluster Blood BA9 BA10 BA8 EntCtx STG Cerebellum Individual differences – conserved across blood and brain? r=0.87, p<0.0001 Much more data needed! Cell-type-specific methylomes: neurons, astrocytes, glia Katie Lunnon, Jon Cooper 5. We may be trying to detect inherently small effect sizes using sub-optimal methods and sample cohorts Do the small DNA methylation differences often observed between groups translate into differences in gene expression in the relevant tissue?? 1500bp upstream of transcription start siteP<1x10-8 P-Value HOXB8 NKX2-5 C17orf100 SGCE C4orf38 HAUS2 LOC399815 LRP1B FAM92B 4.10E-06 2.54E-05 8.10E-05 0.000114176 0.0001863 0.000211401 0.000267029 0.000327942 0.000347768 Mean meth difference=-0.02 Adjusted P-value Methylation difference 0.08 -0.02 0.26 -0.03 0.53 -0.01 0.53 -0.01 0.53 -0.01 0.53 -0.01 0.53 0.04 0.53 0.04 0.53 0.02 Confirming findings in cleaner model systems (e.g. cell and animal models) Control for confounding factors and environmental influences But a mouse is not a man, and a cell-line is not a body 6. We lack a framework for the analysis of genome-wide epigenetic data Reference epigenomes – across cells and tissues – what is normal? Cataloging regions of high inter-individual variation Integrating epigenomic data with genetics and other –omics information Reference Epigenomes Technology Development Novel Epigenetic Marks Epigenomics of Human Health & Disease Neurodegeneration Bipolar disorder Schizophrenia Autism Atherosclerosis Hypertension SLE Kidney disease Asthma Insulin Resistence Click here to browse data http://www.roadmapepigenomics.org Where is the data: sites with unique features Consortium homepage http://roadmapepigenomics.org • View data on genome • protocols • standards NCBI http://ncbi.nlm.nih.gov/epigenomics http://ncbi.nlm.nih.gov/geo/roadmap/epigenomics • View data • Download data • Compare samples Human Epigenome Atlas http://epigenomeatlas.org • View data on genome or with Atlas gene browser • Download data • Tools at Genboree Workbench WashU VizHub http://vizhub.wustl.edu • Next-gen browser http://epigenomegateway.wustl.edu • UCSC visualization hub at http://genome.ucsc.edu What data are available to me? Range of cells/tissues covered: Currently 125 cell/tissue types represented including…. iPS and ES cells, some differentiated forms Fetal tissues (heart, brain, kidney, lung, others) Adult primary cells and tissues (hematopoietic, brain regions, breast cell types, liver, kidney, colon, muscle, adipocytes, others) Most samples will have: DNA methylation data (RRBS, MRE-seq, MeDIP-seq, whole genome bisulfite seq) ChIP-seq data (currently H3K27me3, H3K36me3, H3K4me1, H3K4me3, H3K9me3) DNase I hypersensitivity data Gene expression data (arrays or RNA-seq) Some samples will have: Expanded panel of histone modifications (currently 20+) Can download: .wig, .bed, some .bam, SRA, peak calls Two ways to browse: Data Table Type search terms here to narrow list Search isn’t literal (…type “lung”, “blood”) Two ways to browse: Visual Browser Mouse over sites, click for table Genome-wide epigenetic data at NCBI: Epigenomics Gateway Tutorials Compare samples Text search or browser search http://www.ncbi.nlm.nih.gov/epigenomics Compare Samples: Identify genes with significant epigenetic differences GO Terms and pathways found most frequently The Human Epigenome Atlas Genboree workbench Click for data Click to view selected data sets Click to download data, or for metadata A new epigenomics browser: data and metadata together Click here for browser http://epigenomegateway.wustl.edu significance What can I do with the data: interpret GWAS hits Genomic locus Data from ENCODE: Human and mouse: http://genome.ucsc.edu/ENCODE Fly and worm: http://modencode.org Top SNPs linked to cell type specific enhancer states in disease relevant cell types Ernst et al, Nature 2011 Beyond GWAS: Integrated genetic-epigenetic approach to common disease 7. Back to hype and bad science reporting – especially with regard to “transgenerational epigenetic inheritance” We need to manage expectations Epigenetic profile erased and reset de novo during gametogenesis Transgenerational longitudinal cohort studies Questions? Lecturer in Epigenetics Bioinformatic approaches and computational epigenomics Environmental epigenomics Functional epigenomics Postdoctoral Research Workers Laboratory Technician PhD students Contact: [email protected] www.epigenomicslab.com