Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
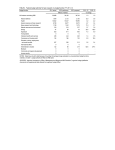
Supplemental Material Common Variant Associations with Fragile X Syndrome TABLE OF CONTENTS Supplemental Methods ..................................................................................................................... 2 Functional genomics ...................................................................................................................................................2 Supplemental Figures ........................................................................................................................ 4 Supplemental Figure 1. Sample ancestry ...................................................................................................................4 Supplemental Figure 2. SNP allele frequencies in FXS cases & controls ....................................................................5 Supplemental Figure 3. Comparison of relatedness in FXS cases & controls ............................................................6 Supplemental Figure 4. Brain RNA-seq Data in FMR1 association region .................................................................7 Supplemental Figure 5. 5C results for the FMR1 region ............................................................................................8 Supplemental Tables ......................................................................................................................... 9 Supplemental Table 1. Subject characteristics ...........................................................................................................9 Supplemental Table 2. Fine-mapping results .......................................................................................................... 10 References ...................................................................................................................................... 11 Page 1 of 12 SUPPLEMENTAL METHODS FUNCTIONAL GENOMICS Brain samples. Dorsolateral prefrontal cortex (Brodmann area 9) dissected from postmortem samples from nine adult schizophrenia cases and nine adult control brains were obtained from Dr Craig Stockmeier (University of Mississippi Medical Center) (1). Cases and controls were sex- and age-matched, and all were of European ancestry. Controls had no history of psychiatric disorders or substance abuse. Frontal cortex from nine fetal brains, gestational age 17-19 weeks, were obtained from the NIH NeuroBiobank (https://neurobiobank.nih.gov). All fetal samples were of African American ancestry. Samples were genotyped on the Illumina Human Omni Express chip in order to confirm sample integrity. Samples were dry homogenized to a fine powder using a liquid nitrogen-cooled mortar and pestle. Aliquots from each sample were prepared, and used for multiple purposes including ATAC-seq, RNA-seq, carbon-copy chromatin conformation capture (5C), and DNA microarray. Sample processing was conducted blind to case-control status. Cell lines. Three neural progenitor cell lines were used: an NIH human iPSC, H1 hESC, and ReNCellVM cells. We used a modified protocol from Zeng et al. (2) to differentiate NIH human iPSCs and H1 hESCs. iPSCs were collected after 51 days of differentiation, and H1 hESCs were collected after 47 days of differentiation. ReNCellVM cells were grown and differentiated according to the pre-aggregation differentiation protocol by Donato et al. (3). RNA-seq. RNA-sequencing data was generated from all brain samples and cell lines. We extracted total RNA from 25 mg of pulverized tissue from fetal and adult brain using Norgen’s Fatty Tissue RNA Purification Kit (Norgen Biotek, Thorold, ON Canada). Extracted RNA from the cell pellets (each with ~3-6 million cells) using Norgen’s Total RNA extraction kit. RNA concentration was measured using fluorometry (Qubit 2.0 Fluorometer), and RNA quality verified using a microfluidics platform (Bioanalyzer, Agilent Technologies). Barcoded libraries were created (Illumina TruSeq RNA Sample Preparation Kit v4) using 1 µg of total RNA as input. Samples were randomly assigned to bar codes and lanes. Libraries were quantified using fluorometry and equal amounts of all barcoded samples were pooled and sequenced using Illumina HiSeq 2000 (100 bp paired-end reads). We mapped lane-level reads to the human genome (hg19) with Tophat (4) (v2.0.6) using default parameters and the --library-type fr-firststrand option for strand-specific libraries. Using samtools (5), we removed reads with quality score <10 or potential PCR duplicates. Mapped reads were summarized into gene-level expression estimates of total read count (TReC). TReC is the number of reads that overlapped exonic regions of a gene (determined using countReads in the R package isoform (6)). Gene models were based on non-overlapping exons from Ensembl. genes. We excluded genes with very low expression levels (sum of TReC across all samples <50). TReC data were normalized using the weighted trimmed mean of M-values scale-normalization method in EdgeR (7). Total-stranded RNA-seq. We used total-stranded RNA-seq to detect unannotated features on five samples: DLPFC from a control, PFC from a fetal sample, and three neural progenitor cell lines (differentiated NIH human iPSCs, H1 hESCs, and ReNCellVM cells). RNA was extracted as detailed above. 5C. Chromosome conformation capture-carbon copy (5C) was performed on five human fetal prefrontal cortex samples. The region targeted by 5C was chrX:146200000-147460000 (hg19). Tissue processing was based on two previously published protocols (8, 9), one of which was designed for postmortem brain tissue. (9). Frozen tissue (~250 mg) was pulverized using the Cellcrusher. To produce a single cell suspension, pulverized tissue was treated with 1mL of 0.125% (w/v) collagenase in 1x PBS and placed in shaker/incubator (300 RPM, 30 minutes, Page 2 of 12 37°C). Tissue was then Dounce homogenized with pestle A (10x) and returned to shaker/incubator (300 RPM, 30 minutes, 37°C) followed by further dissociation by Dounce homogenization with pestle B (10x), and spun at 400xg for 2 minutes at room temperature. After discarding the supernatant, the pellet was be re-suspended in 2 mL of 1x PBS. Cells were filtered using a 40 µm cell strainer, and transferred to a 15 mL conical tube. Following confirmation of single cell suspension and quantification of cells (Countess Automatic Cell Counter), we proceeded with the 5C protocol. We crosslinked cells with formaldehyde (1% final concentration) and quenched the reaction using glycine (150 mM). Cells were then lysed and DNA was digested using HindIII. 3C library preparation were done according to protocol by Naumova et al. (10) 5C library preparation was done according to protocol by van Berkum et al. (11) 5C primers (226 forward primers; 6 reverse primers) were designed using the my5C.primers module from the my5C web tools for 5C experiments and based on hg19. (12) Reverse primers were based on predicted transcription start sites (TSS; Gencode V17 Comprehensive), while forward primers mapped to all other HindIII fragments, with acceptable primers according to specific criteria from my5C.primers module. 5C libraries were indexed (KAPA Hyper) and sequenced on two lanes of Illumina HiSeq2500 (2x100bp). Plots were generated with the Bioconductor package Sushi. Two criteria for interactions were examined: a liberal criterion (FDR < 0.1 in 3 of 5 samples) and a more conservative criterion (FDR < 0.05 in 4 of 5 samples). ATAC-seq library preparation and sequencing. Assay for transposase-accessible chromatin sequencing (ATACseq) was used to map chromatin accessibility genome-wide. This method probes DNA accessibility with hyperactive Tn5 transposase, which inserts sequencing adapters into accessible regions of chromatin. We used tissue from human DLPFC of nine schizophrenia cases and nine controls along with PFC from nine fetuses (see above for sample description). Approximately 20 mg of pulverized material was used for ATAC-seq. Frozen samples were thawed in 1 ml of nuclear isolation buffer (20 mM Tris-HCL, 50 mM EDTA, 5mM Spermidine, 0.15 mM Spermine, 0.1% mercaptoethanol, 40% glycerol, pH 7.5), inverted for 5 minutes to mix, and samples were filtered through Miracloth to remove larger pieces of tissue. Samples were centrifuged at 1100 x g for 10 min at 4°C. The resulting pellet was washed with 50 µl RSB buffer, centrifuged again, and supernatant was removed. The final crude nuclear pellet was re-suspended in transposition reaction mix and libraries prepared for sequencing as described in Buenrostro et al. (13) All samples were barcoded, and combined into pools. Each pool contained 6 randomly selected samples. Each pool was sequenced on two lanes of an Illumina 2500 sequencer (San Diego, CA, USA). The raw fastq files were processed through cutadapt (version 1.2.0, URLs) (14) to remove adaptors and lowquality reads. cutadapt-filtered reads were aligned to hg19 using bowtie2 (version 2.1.0, URLs) (15) using default parameters. In alignment, all reads are treated as single-read sequences, regardless of whether ATAC-seq libraries were sequenced as single-end or paired-end. The aligned bam files were sorted using samtools (version 0.1.18, URLs), (16) duplicates removed using Picard MarkDuplicates (URLs), and then converted to bed format using BedTools (version: v2.17.0, URLs). (17) ENCODE “blacklist” regions were removed (i.e., empirically identified genomic regions that produce artefactual high signal in functional genomic experiments). Narrow open chromatin peaks were called from the final bed files using MACS2 (URLs). For visualization, bigwig files were generated using wigToBigWig (version 4, URLs) (18) and bedgraph files were output by MACS2. Page 3 of 12 SUPPLEMENTAL FIGURES SUPPLEMENTAL FIGURE 1. SAMPLE ANCESTRY Scatterplot of the first two principal components (PCs) for FXS cases (filled diamonds), GABC controls (open squares), and the means for ten HapMap 3 samples. Nine FXS cases are outliers, eight with variable African ancestry and one that clustered with the GIH / MEX HapMap samples. Abbreviations. ASW=African ancestry in Southwest USA. CEU=Utah residents with Northern and Western European ancestry from the CEPH collection. CHB=Han Chinese in Beijing, China. CHD=Chinese in Metropolitan Denver, Colorado. GIH=Gujarati Indians in Houston, Texas. JPT=Japanese in Tokyo, Japan. LWK=Luhya in Webuye, Kenya. MEX=Mexican ancestry in Los Angeles, California. TSI=Toscani in Italia. YRI=Yoruba in Ibadan, Nigeria. Page 4 of 12 SUPPLEMENTAL FIGURE 2. SNP ALLELE FREQUENCIES IN FXS CASES & CONTROLS Scatterplot of the allele frequencies in FXS cases and control for all GWAS SNPs. Contours indicate SNP density. Most SNPs had similar allele frequencies in cases and controls (Spearman =0.97, P <10-300). Five SNPs (all in the vicinity of FMR1, red X’s) had divergent allele frequencies, and reached genome-wide significance in case-control analyses (P<5x10-8). Page 5 of 12 SUPPLEMENTAL FIGURE 3. COMPARISON OF RELATEDNESS IN FXS CASES & CONTROLS Genomic relatedness was computed for all pairs of FXS and control subjects using autosomal, LD-pruned SNPs. Subject quality control had previously removed one member of any pair of subjects with 𝜋̂ > 0.20. Three pairs of cases (6 individuals) were related at approximately the third-degree. Despite this, the median 𝜋̂ values were lower for case-case pairs (0.0078) than control-control pairs (0.0094). In other words, case pairs tended to be less interrelated than control pairs. Page 6 of 12 SUPPLEMENTAL FIGURE 4. BRAIN RNA-SEQ DATA IN FMR1 ASSOCIATION REGION The top tracks show GENCODE genes, classical FMR1 markers, and the fine-mapping results. The next track is empty as there was no evidence of RNA alignments to genomic areas without current annotations. The next tracks show gene-level expression in brain from RNA-seq of dorsolateral prefrontal cortex (DLPFC) in 9 SCZ cases and 9 controls along with prefrontal cortex (PFC) from 9 fetuses. FMR1 was robustly expressed, but there was no detectible expression of FMR1-AS1. We used total-stranded RNA-seq to detect unannotated features on 5 samples: DLPFC in a control, PFC in a fetal sample, and three neural progenitor cell lines (an NIH human IPSC on day 51 of a differentiation protocol, H1 cells on day 47 of a differentiation protocol, and ReNCellVM cells). All samples had substantial alignments to the forward strand (blue) mostly corresponding to known FMR1 exons. The reverse strand alignments (red) had very low counts consistent with “leaky” reverse direction transcription from the FMR1 promoter. We did not find evidence of an unannotated feature in the association region. Page 7 of 12 SUPPLEMENTAL FIGURE 5. 5C RESULTS FOR THE FMR1 REGION 5C (chromosome conformation capture-carbon copy) results from five human fetal prefrontal cortex samples. The region shown was targeted by 5C (hg19, chrX:146200000-147460000). Plots were generated with the Bioconductor package Sushi. The top plot shows interactions with a liberal criterion (FDR < 0.1 in 3 of 5 samples), and the middle plot a more conservative criterion (FDR < 0.05 in 4 of 5 samples). The bottom track shows GENCODE genes, the positions of classical markers (including the FMR1 promoter CGG repeat), and the genome-wide significant common variation. The miRNA gene cluster on the left was unremarkable in the GWAS, but has dense DNA-DNA interactions, including some with the GWAS region. Interaction involving the FMR1 promoter containing the CGG repeat are discussed in the text. Page 8 of 12 SUPPLEMENTAL TABLES SUPPLEMENTAL TABLE 1. SUBJECT CHARACTERISTICS Characteristic FXS Cases Controls 89 266 100% 100% Mean age at assessment (SD) 10.5 (5.2) 20.8 (3.5) Vineland adaptive behavior scale, socialization score, mean (SD) 53.8 (17.0) – Vineland adaptive behavior scale, composite score, mean (SD) 43.1 (14.9) – Number Percent male SD=standard deviation. Vineland scores were standardized to mean=100 and SD=15. FXS cases were markedly impaired. Page 9 of 12 SUPPLEMENTAL TABLE 2. FINE-MAPPING RESULTS chrX (hg19) SNP Alleles Fcase Fcontrol P (fine mapping) OR SE 146844358 P (Table 2) rs73602793 A/C 0.09574 0.05139 0.095 1.954 0.4084 146852679 rs5952060 C/T 0.7835 0.4004 5.91e-12 5.419 0.264 146853379 rs141685640 G/A 0.03093 0.0364 0.79 0.8448 0.6364 146861015 rs45631655 A/T 0.4124 0.1799 4.88e-7 3.2 0.2389 146865083 rs148756788 C/T 0.1957 0.1073 0.018 2.024 0.3024 146876718 rs112146098 C/G 0.3505 0.1156 6.609e-9 4.128 0.2573 146882730 rs61577930 G/A 0.06186 0.06852 0.81 0.8963 0.4596 146884909 rs112866660 C/T 0.02062 0.03212 0.55 0.6344 0.7612 146886586 rs5905137 C/T 0.5567 0.2805 1.35e-7 3.221 0.2289 146895120 rs2197706 A/C 0.8041 0.3876 6.83e-14 6.487 0.2729 2.53e-10 146908213 rs5905149 A/C 0.6289 0.2655 4.04e-12 4.687 0.2348 5.76e-10 146908857 rs12009090 T/C 0 0.002169 0.65 0 146913828 rs7876251 G/A 0.7113 0.3362 6.85e-12 4.866 0.2445 3.68e-9 146918268 rs4824253 G/A 0.7113 0.3362 6.85e-12 4.866 0.2445 6.41e-9 146923549 rs5905152 C/T 0.01031 0 0.028 146925273 rs138345743 A/T 0.01042 0.01293 0.84 0.8035 1.086 146939662 rs45631657 C/T 0.375 0.0991 5.20e-12 5.455 0.264 146940339 rs139987830 A/C 0 0.002141 0.65 0 146944900 rs12008373 T/A 0 0.002141 0.65 0 146951567 rs148850319 C/T 0 0.002141 0.65 0 146955690 rs149005990 A/G 0 0.002146 0.65 0 146962305 rs140441434 T/C 0 0.002141 0.65 0 146963621 rs4824231 T/C 0.7423 0.3212 1.13e-14 6.086 146968287 rs181526497 A/T 0 0.03426 0.064 0 146980516 rs25705 T/C 0.433 0.167 5.74e-9 3.808 0.2395 146989460 rs12556135 T/C 0.07216 0.1159 0.21 0.5934 0.4182 146994239 rs146302130 C/G 0 0.02141 0.15 0 146994959 rs1805422 C/G 0.04124 0.0985 0.071 0.3936 146995450 rs12013181 C/T 0 0.002141 0.65 0 147003339 rs25726 G/A 0.06186 0.1113 0.14 0.5262 0.4464 147010529 rs25708 A/G 0.05155 0.02784 0.23 1.898 0.5385 147013704 rs29283 G/A 0 0.002151 0.65 0 4.57e-8 0.2524 0.5337 Fine-mapping using 32 SNPs in 97 FXS cases and 467 male controls. The first allele given is the least common in this sample and is the reference for the odds ratio (OR) and frequencies in male cases (Fcase) and controls (Fcontrol). SE is standard error. The first P is for fine mapping, and the second is from the initial GWAS. Genome-wide significant SNPs are highlighted in red. Page 10 of 12 REFERENCES 1. Zhu H, Urban DJ, Blashka J, McPheeters MT, Kroeze WK, Mieczkowski P, et al. Quantitative analysis of focused A-to-I RNA editing sites by ultra-high-throughput sequencing in psychiatric disorders. PLoS ONE. 2012;7(8):e43227. 2. Zeng H, Guo M, Martins-Taylor K, Wang X, Zhang Z, Park JW, et al. Specification of region-specific neurons including forebrain glutamatergic neurons from human induced pluripotent stem cells. PLoS ONE. 2010;5(7):e11853. 3. Donato R, Miljan EA, Hines SJ, Aouabdi S, Pollock K, Patel S, et al. Differential development of neuronal physiological responsiveness in two human neural stem cell lines. BMC Neurosci. 2007;8:36. 4. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105-11. 5. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078-9. 6. Sun W, Liu Y, Crowley JJ, Chen TH, Zhou H, Chu H, et al. IsoDOT Detects Differential RNA-isoform Expression/Usage with respect to a Categorical or Continuous Covariate with High Sensitivity and Specificity. J Am Stat Assoc. 2015;110(511):975-86. 7. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139-40. 8. Stadhouders R, Kolovos P, Brouwer R, Zuin J, van den Heuvel A, Kockx C, et al. Multiplexed chromosome conformation capture sequencing for rapid genome-scale high-resolution detection of long-range chromatin interactions. Nat Protoc. 2013;8(3):509-24. 9. Mitchell AC, Bharadwaj R, Whittle C, Krueger W, Mirnics K, Hurd Y, et al. The genome in three dimensions: a new frontier in human brain research. Biol Psychiatry. 2014;75(12):961-9. 10. Naumova N, Smith EM, Zhan Y, Dekker J. Analysis of long-range chromatin interactions using Chromosome Conformation Capture. Methods. 2012;58(3):192-203. 11. van Berkum NL, Dekker J. Determining spatial chromatin organization of large genomic regions using 5C technology. Methods Mol Biol. 2009;567:189-213. 12. Lajoie BR, van Berkum NL, Sanyal A, Dekker J. My5C: web tools for chromosome conformation capture studies. Nat Methods. 2009;6(10):690-1. 13. Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10(12):1213-8. 14. Martin M. Cutadapt Removes Adapter Sequences From High-Throughput Sequencing Reads. EMBnetjournal.17. 15. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357-9. Page 11 of 12 16. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078-9. 17. Quinlan AR. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr Protoc Bioinformatics. 2014;47:11 2 1-34. 18. Speir ML, Zweig AS, Rosenbloom KR, Raney BJ, Paten B, Nejad P, et al. The UCSC Genome Browser database: 2016 update. Nucleic Acids Res. 2016;44(D1):D717-25. Page 12 of 12