Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Adenosine triphosphate wikipedia , lookup
Ribosomally synthesized and post-translationally modified peptides wikipedia , lookup
Clinical neurochemistry wikipedia , lookup
Nitrogen cycle wikipedia , lookup
Nucleic acid analogue wikipedia , lookup
Butyric acid wikipedia , lookup
Catalytic triad wikipedia , lookup
Point mutation wikipedia , lookup
Metalloprotein wikipedia , lookup
Fatty acid metabolism wikipedia , lookup
Proteolysis wikipedia , lookup
Fatty acid synthesis wikipedia , lookup
Glyceroneogenesis wikipedia , lookup
Protein structure prediction wikipedia , lookup
Peptide synthesis wikipedia , lookup
Citric acid cycle wikipedia , lookup
Genetic code wikipedia , lookup
Biochemistry wikipedia , lookup
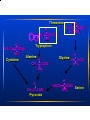
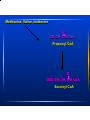
Amino Acid Oxidation and the Urea Cycle Amino Acids: • Final class of biomolecules whose oxidation contributes significantly to the generation of energy • Undergo oxidation in three metabolic circumstances in animals: During normal synthesis and degradation of proteins When a diet is rich in protein; amino acids are not stored During starvation or in diabetes mellitus Overview of Amino Acid Catabolism: Dietary protein Intracellular protein Glucose TCA Cycle Respiration α-keto acid Amino Acids NH44++ Biosynthesis of amino acids nucleotides, biological amines Excretion Metabolic Fates of Amino Groups Most amino acids are metabolized in the liver • Some of the ammonia generated is used in biosynthesis; the excess is excreted • Excess ammonia generated in extrahepatic tissues is transported to liver for conversion to the appropriate excreted form Removal Removal of of the the α-amino α-amino groups groups occurs occurs in in the the cytosol cytosol by by transamination transamination reactions reactions catalyzed catalyzed by by aminotransferases aminotransferases (transaminases): (transaminases): α-keto amino acid acid11 α-keto acid acid22 ++ amino α-keto α-keto acid acid11 ++ amino amino acid acid22 In In liver liver α-keto α-keto acid acid11 is is usually usually α-Kg; α-Kg; in in muscle muscle it it is is usually usually pyruvate pyruvate COO-- COO-COO-- C=O + CH22 CH22 COO-α-Kg -- COO C=O CH22 CH22 COO-α-Kg + ++ 33 NH H33N++ C H R amino acid COO-H33N++ C H CH33 alanine C-H CH22 + CH22 COO-glutamate GPT alanine alanine aminoamino- COO-NH33++ C-H CH22 + CH22 transferase transferase COO-glutamate COO-C=O R α-keto acid COO-C=O CH33 pyruvate COO-C=O CH22 CH22 COO-α-Kg -- + COO H33N++ C H CH22 COO-aspartate COO-NH33++ C-H CH22 + CH22 COO-- COO-C=O CH22 COO-OAA glutamate Aspartate aminotransferase or Glutamate-OAA transaminase (GOT) Serum Serum GPT GPT (SGPT) (SGPT) and and GOT GOT (SGOT) (SGOT) are are sensitive sensitive indicators indicators for for aa number number of of disease disease conditions. conditions. • During heart attacks, damaged heart cells leak aminotransferases. • Damaged liver cells also leak aminotransferases. SGPT and SGOT levels are monitored in people exposed to industrial chemicals. The The effect effect of of transamination transamination is is to to collect collect amino amino groups groups from from many many amino amino acids acids and and convert glutamate convert them them into into one, one, glutamate • Glutamate channels amino groups into biosynthetic pathways or into reactions where nitrogenous waste products are formed How How are are amino amino groups groups removed removed from from glutamate glutamate and and prepared prepared for for excretion? excretion? Glutamate is transported into the mitochondrial matrix where it undergoes oxidative deamination catalyzed by glutamate d’hase: Glutamate glutamate d’hase NAD(P)+ NAD(P)H α-Kg + NH44 Ammonia Ammonia is is extremely extremely toxic toxic to to animal animal tissues; tissues; it it is is converted converted to to glutamine glutamine for for transport transport from from extrahepatic extrahepatic tissues tissues to to the the liver liver or or kidneys. kidneys. COO-- COO-NH33++ C-H CH22 CH22 COO-glutamate glutamine synthetase NH44 NH33++ C-H CH22 CH22 H22N C=O glutamine In In the the liver liver glutamine glutamine is is converted converted back back to to glutamate glutaminase: glutamate by by glutaminase: COO-NH33++ C-H CH22 CH22 H22N C=O COO-glutaminase H22O NH44++ glutamine UREA NH33++ C-H CH22 CH22 COO-glutamate • Glutamine is the major transport form of ammonia; it is present in blood in much higher concentrations than other amino acids. • Alanine also plays a role in transport of amino groups to the liver by the glucose-alanine cycle: Muscle AA glutamate Liver glucose pyr + glutamate glutamate pyr alanine + α−Kg α−Kg alanine Habitat Habitat determines determines the the Molecular Molecular Pathway Pathway for for Nitrogen Nitrogen Excretion Excretion • Aquatic organisms (bacteria, protozoa, fish) release ammonia to their aqueous enviroment (ammonotelic) • Birds and reptiles convert amino nitrogen into uric acid; they cannot carry enough water for the excretion of nitrogen as urea (uricotelic) • Terrestrial animals excrete amino nitrogen in the form of urea (ureotelic) • Plants recycle virtually all amino groups; there is no general pathway for nitrogen excretion The Urea Cycle • Discovered by Hans Krebs and Kurt Hanseleit • Occurs in the liver • Takes place in two intracellular compartments; the cytosol and the mitochondrial matrix ++ 44 NH + carbamoyl phosphate HCO33-synthetase I 2 ATP 2 ADP, Pi Pi NH33++ O H22N-C-NH-(CH22)33-CH-COO-- O O H22N-C-O-P-O-O-carbamoyl phosphate ornithine ornithine transcarbamoylase transcarbamoylase NH33++ NH33-(CH22)33-CH-COO-Ornithine Citrulline Transported to cytosol NH33++ O H22N-C-NH-(CH22)33-CH-COO-Citrulline Aspartate arginosuccinate synthetase ATP ATP PPi PPi NH33++ COO-- NH22++ -OOC-CH22-CH-NH-C-NH-(CH22)33-CH-COO-Arginosuccinate NH33++ COO-- NH22++ -OOC-CH22-CH-NH-C-NH-(CH22)33-CH-COO-Arginosuccinate arginosuccinate lyase OOC-CH=CH-COO-- -- Fumarate NH33++ NH22++ -) -CH-COO NH2-C-NH-(CH 22 33 2 Arginine NH33++ NH22++ -) -CH-COO NH2-C-NH-(CH 22 33 2 Arginine arginase NH33++ NH33-(CH22)33-CH-COO-Ornithine O H22N-C-NH22 UREA Back to mitochondrion Overall equation for Urea synthesis: NH33 + HCO33-- + Aspartate + 3 ATP urea + fumarate + 2ADP + 2Pi + AMP + PPi Regulation of the Urea Cycle: • Carbamoyl Phosphate Synthetase I is allosterically activated by N-acetylglutamate. • High levels of transamination during amino acid breakdown lead to elevated glutamate with concommitant increases in the concentration of N-acetylglutamate . Breakdown of Individual Amino Acids Degradation of the carbon skeletons of the 20 common amino acids yields one of 7 intermediates: a-Kg, succinyl CoA, pyruvate, fumarate, OAA, acetoacetate, acetyl CoA α-Kg, succinyl CoA, pyruvate, fumarate, OAA can all serve as precursors for glucose synthesis; hence amino acids giving rise to these intermediates are Glucogenic. Acetoacetate and acetyl CoA can serve as precursors for fatty acid or ketone synthesis; hence amino acids giving rise to these compounds are termed Ketogenic Alanine, Serine, Glycine, Cysteine, Threonine, Tryptophan Pyruvate Isoleucine, Leucine, Threonine, Tryptophan Acetyl CoA Leucine, Lysine, Phenylalanine, Tyrosine Acetoacetate Arginine, Glutamine, Glutamate, Proline, Histidine Isoleucine, Methionine, Valine Aspartate, Tyrosine, Phenylalanine Aspartate, Asparagine α−Kg Succinyl CoA Fumarate OAA Threonine H CH22 C-COO-NH22 NH Tryptophan H HS-CH22C-COO-NH22 Alanine H Cysteine CH33 C-COO-NH22 O CH33-C-COO-Pyruvate HH H33C-C-C-COO-HO NH22 H Glycine H C-COO-NH22 H HOCH22 C-COO-Serine NH22 • Alanine is converted to pyruvate by transamination. • Asparagine is converted to Aspartate by Asparaginase: H C-CH22 C-COO-NH22 NH22 O H C-CH22 C-COO--O NH22 O • Aspartate can be converted to OAA by transamination: Aspartate + α-KG • Aspartate degradation via the Glutamate + OAA urea cycle yields fumarate. Methionine, Valine, Isoleucine O CH33-CH22-C-S-CoA Propionyl CoA O -OOC-CH22-CH22-C-S-CoA Succinyl CoA Arginine, Glutamine, Histidine, Proline Glutamate Glutamate D’hase α−Kg Leucine Leucine and and Lysine Lysine are are the the only only two two purely purely ketogenic ketogenic amino amino acids. acids. HMG-CoA HMG-CoA is is an an intermediate intermediate in in leucine leucine degradation. degradation. • Initial steps in valine, leucine, isoleucine degradation are identical: transamination to the corresponding a-keto acid decarboxylation to the corresponding CoA derivative dehydrogenation to form a double bond Defect in the decarboxylation reaction results in Maple Syrup Urine Disease, which is fatal unless treated early in life. Phenylalanine is converted to Tyrosine by Phenylalanine Hydroxylase; then the pathway proceeds with the breakdown of tyrosine to fumarate plus acetoacetate. Homogentisate is an intermediate in this pathway. Alkaptonuria results in the urinary excretion of excess homogentisate; air oxidation causes this compound to turn dark. This disease is not fatal, individuals tend to suffer arthritis in later life. OH -OOC-H22C homogentisate OH Phenylketonuria results from absence of phenylalanine hydroxylase. Phe is converted to phenylpyruvate and excreted. Severe mental retardation occurs unless infants are immediately placed on a diet low in Phe. CH22 C=O COO- phenylpyruvate Human Human Genetic Genetic Disorders Disorders of of Amino Amino Acid Acid Catabolism Catabolism Incidence Condition (per (per 100,000 100,000 births) births) Defective Enzyme Symptoms Dark Dark pigment pigment in in urine; urine; arthritis arthritis in in late late life life Alkaptonuria Alkaptonuria 0.4 0.4 Homogentisate Homogentisate Dioxygenase Dioxygenase Maple Maple Syrup Syrup Urine Urine Disease Disease 0.4 0.4 Branched Mental retardation; retardation; Branched chain chain α-keto α-keto Mental acid convulsions; early early acid dedydrogenase dedydrogenase convulsions; death death Phenylketonuria Phenylketonuria Methylmalonic Methylmalonic Acidemia Acidemia (MMA) (MMA) 88 (<0.5) (<0.5) phenylalanine phenylalanine hydroxylase hydroxylase Neonatal Neonatal vomiting; vomiting; mental mental retardation retardation Methylmalonyl Methylmalonyl CoA CoA Mutase Mutase Mental Mental retardation; retardation; convulsions; convulsions; early early death death