Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Artificial gene synthesis wikipedia , lookup
Gene therapy wikipedia , lookup
History of genetic engineering wikipedia , lookup
Nutriepigenomics wikipedia , lookup
Gene expression programming wikipedia , lookup
Genetic engineering wikipedia , lookup
Genetic drift wikipedia , lookup
Gene desert wikipedia , lookup
Genealogical DNA test wikipedia , lookup
Genome evolution wikipedia , lookup
Human genome wikipedia , lookup
Heritability of IQ wikipedia , lookup
Neuronal ceroid lipofuscinosis wikipedia , lookup
Dominance (genetics) wikipedia , lookup
Molecular Inversion Probe wikipedia , lookup
Medical genetics wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Epigenetics of neurodegenerative diseases wikipedia , lookup
Population genetics wikipedia , lookup
Behavioural genetics wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Microevolution wikipedia , lookup
Genome (book) wikipedia , lookup
Designer baby wikipedia , lookup
Human genetic variation wikipedia , lookup
SNP genotyping wikipedia , lookup
Public health genomics wikipedia , lookup
A30-Cw5-B18-DR3-DQ2 (HLA Haplotype) wikipedia , lookup
Haplogroup G-M201 wikipedia , lookup
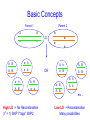
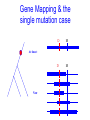
Trait Mapping •Recombination Mapping •SNP mapping BIO520 Bioinformatics Jim Lund Why do we care about variations? underlie phenotypic differences cause inherited diseases allow tracking human history (ancient and modern) Traits • Mendelian – single locus, few alleles – high penetrance, high expressivity – eg color, enzyme, molecular, genetic diseases (CF, hemophilia…) • Quantitative – – – – multiple allele, multilocus variable penetrance, expressivity epistasis, environmental effects eg. blood pressure, weight, IQ... Traits How do we find their basis? • Association of variance in trait with variance in gene • Genetic linkage Basic Concepts Parent 2 Parent 1 A B a b A X B a b A B a b A B a b OR a b A B A B a b High LD -> No Recombination (r2 = 1) SNP1 “tags” SNP2 A b A B a B A B A b a B A B A b etc… Low LD -> Recombination Many possibilities Mapping Issues • Need many arbitrary, polymorphic markers for dense map – Molecular markers: RFLP, STS, SNP • Need many progeny – 100 progeny for 1 cM map – 1000/0.1 cM map, 100 kb in mouse • Map distance varies (the ratio of kb/cM not constant) – centromere suppression – inversion suppression Genetic crosses • Model organisms, e.g. Fungi, no problem • Humans – rare woman who will bear >5, >10 children – controlled breeding problematic Alternate Mapping • Pedigree analyses – likelihood estimation – The original method, now less common • Population-based mapping – association studies – linkage disequilibrium Pedigree Analysis • Likelihood Method (LOD scores) • LOD 3-4, 1/1000 – 1/10000 odds of linkage – genome-wide p-value of p < .05 • Hard to extend to <1 cM Cloning Human Genes • • • • Positional Positional/Candidate Candidate Only Functional Complex diseases Association mapping • Disease gene: D, d • Marker: M, m M associated with D if the probability of an individual having the disease given that they have allele M is much greater than the chance of having the disease if the individual has allele m. Written as: P(D|M) > P(D|m) Linkage between the gene and marker increases the likelihood of D M1 M2 M3 M4 M5 M6 association. Association can be caused by – – – – Causation Population subdivision Statistical artifact Linkage disequilibrium Association Mapping •Pedigree sampled •Many Meiosis (>104) •Limited by number of markers M r D 2N generations •Resolution: 10-5 Morgans (Kbases) Gene Mapping & the single mutation case D M At time t D Now M Complicating factors Major Disease Causing Mutation. Minor Disease Causing Mutation + + has the disease. + + Non-genetic cause + + + Incomplete penetrance Oversampled Alzheimers & Apolipoproteins E Definition of QTL? A quantitative trait locus (QTL) is the location of individual or multiple loci that affects a trait that is measured on a quantitative (linear) scale. Examples of quantitative traits are blood pressure and grain yield (measured on a balance). These traits are typically affected by more than one gene, and also by the environment. Thus, mapping QTL is not as simple as mapping a single gene that affects a qualitative trait (such as an inborn error of metabolism). http://gnome.agrenv.mcgill.ca/tinker/pgiv/whatis.htm QTLs-interesting traits • Heritability often ~0.5 • Traits like: – Heart disease – Depression – Type II diabetes – High blood pressure – Arthritis – Most diseases! QTLs-simple problems • 30,000 markers – P-value=0.01 – 299 false hits, 1 real one – Correct for multiple testing • 2 QTLS near one another – “ghost” QTL between them Factors that lead to success in mapping QTLs • Simple, easily quantified trait • Genes of major effect – distinct chromosomal loci • Well-defined map • Large numbers of progeny – inbred – outbred Significance Thresholds by Permutation Churchill and Doerge, 1994 1.Permute the data (create the null hypothesis) H0: there is no QTL in the tested interval H1: there is QTL in the tested interval 2.Perform interval mapping 3. Repeat (1) and (2) many times 4.Choose Threshold Human SNPs • About 10 million SNPs exist in human populations where the rarer SNP allele has a frequency of at least 1%. • A set of associated SNP alleles in a region of a chromosome is called a "haplotype". • SNPs are arranged in groups – SNPs within groups show little recombination – Nonrandom association of SNPs results in only a few common haplotypes – Patterns capture most of the variation in a region • The HapMap will describe the common patterns of genetic variation in humans. • The HapMap Project will identify the associations between SNPs and identify the SNPs that tag them (tagSNPs). SNPs identification methods • Pairwise sequence comparison • Deep resequencing • High throughput mismatch detection methods – Denaturing high-performance liquid chromatography (DHPLC) – Single-strand Conformational Polymorphism (SSCP) HapMap • Blocks of adjacent SNPs that show little recombination are called haplotype blocks. • Mean haplotype block length is tens of kb. • HapMap project started examining 270 individuals from 4 ethnic groups. • Now expanding to a more comprehensive sample. Characterization of haplotype blocks means that fewer SNPs will need to be typed. 500,000 SNPs will identify 90% of haplotype blocks. HapMap Glossary • LD (linkage disequilibrium): For a pair of SNP alleles, it’s a measure of deviation from random association (i.e., a measure of lack of recombination). Measured by D’, r2, LOD • Phased haplotypes: Estimated distribution of SNP alleles. Alleles transmitted from Mom are in same chromosome haplotype, while Dad’s form the paternal haplotype. • Tag SNPs: Minimum SNP set to identify a haplotype. r2= 1 indicates two SNPs are redundant, so each one perfectly “tags” the other. HapMap Project Phase 1 Phase 2 Phase 3 Samples & POP panels 269 samples (4 panels) 270 samples (4 panels) 1,115 samples (11 panels) Genotyp ing centers HapMap Internati onal Consorti um Perlege n Broad & Sanger Unique QC+ 1.1 M 3.8 M (phase 1.6 M (Affy 6.0 Phase 3 Samples label ASW* CEU* CHB CHD GIH JPT LWK MEX* MKK * TSI YRI* population sample African ancestry in Southwest USA Utah residents with Northern and Western European ancestry from the CEPH collection Han Chinese in Beijing, China Chinese in Metropolitan Denver, Colorado Gujarati Indians in Houston, Texas Japanese in Tokyo, Japan Luhya in Webuye, Kenya Mexican ancestry in Los Angeles, California Maasai in Kinyawa, Kenya Toscans in Italy Yoruba in Ibadan, Nigeria * Population is made of family trios # samples 90 QC+ Draft 1 71 180 162 90 100 100 91 100 90 180 100 180 1,301 82 70 83 82 83 71 171 77 163 1,115 SNP databases • dbSNP (NCBI) – 12 million human SNPs – 5 million validated SNPs – http://www.ncbi.nlm.nih.gov/SNP/get_html.cgi?whichHtml=overview • SNP frequency information • Mapped to the current genome build • HapMap (haplotypes) How to use markers to find disease? genome-wide, dense SNP marker map • problem: genotyping cost precludes using millions of markers simultaneously for an association study • question: how to select from all available markers a subset that captures most mapping information (marker selection, marker prioritization) • depends on the patterns of allelic association (haplotypes) in the human genome The promise for medical genetics CACTACCGA CACGACTAT TTGGCGTAT • within blocks a small number of SNPs are sufficient to distinguish the few common haplotypes significant marker reduction is possible chromosome blocks • if the block structure is a general feature of human variation structure, whole-genome association studies will be possible at a reduced genotyping cost • this motivated the HapMap project Gibbs et al. Nature 2003 The promise for medical genetics •Discover genes contributing to complex diseases •Use these markers to test for inherited disease risk • Find SNPs associated with drug side effects •Make drugs safer. •Rescue drugs abandoned due to significant side effects. Pathway of Drug Development • Lead or Target (Clinical Candidate) • Animal Model Testing – Toxicity, Efficacy • Phase I Pre-Clinical (toxicity) • Phase II (efficacy) • Phase III (efficacy) • NDA (new drug application) • $100M 2000 • $0.5M 100 • $0.5M • $5M • $50M 20 3 2 1 Why pharmacogenomics? • Where do you find the next profitable drug? – The 19/20 drugs that failed AFTER phase 1, but are still efficacious! • How do you decrease the cost of clinical trials? – Don’t enroll people of the “wrong” genotype! • Only give drugs to patients likely to benefit and at a low genetic risk of side effects!