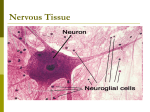
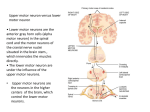
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
List of Papers This thesis is based on the following papers, which are referred to in the text by their Roman numerals. I II III IV Enjin A, Rabe N, Nakanishi ST, Vallstedt A, Gezelius H, Memic F, Lind M, Hjalt T, Tourtellotte WG, Bruder C, Eichele G, Whelan PJ, Kullander K (2010) Identification of novel spinal cholinergic genetic subtypes disclose Chodl and Pitx2 as markers for fast motor neurons and partition cells. J Comp Neurol 518:2284-2304. Wootz H, Enjin A, Wallen-Mackenzie Å, Lindholm D, Kullander K (2010) Reduced VGLUT2 expression increases motor neuron viability in Sod1G93A mice. Neurobiol Dis 37:58-66 Enjin A, Leao KE, Mikulovic S, Le Merre P, Tourtellotte WG, Kullander K. 5-ht1d marks gamma motor neurons and regulates development of sensorimotor connections Manuscript Enjin A, Leao KE, Eriksson A, Larhammar M, Gezelius H, Lamotte d’Incamps B, Nagaraja C, Kullander K. Development of spinal motor circuits in the absence of VIAAT-mediated Renshaw cell signaling Manuscript Reprints were made with permission from the respective publishers. Cover illustration Carousel by Sasha Svensson Contents Introduction.....................................................................................................9 Background ...................................................................................................11 Neural control of movement.....................................................................11 The motor neuron .....................................................................................12 Organization of motor neurons............................................................14 Motor neuron subtypes ........................................................................14 Development of motor neurons ...........................................................16 Degeneration of motor neurons in ALS...............................................17 Proprioception ..........................................................................................18 Anatomy and physiology of the muscle spindle..................................18 Proprioceptive circuits .........................................................................20 Development of the monosynaptic stretch reflex ................................21 Recurrent inhibition..................................................................................22 Development of recurrent inhibition ...................................................23 Aims..............................................................................................................24 Methodological considerations .....................................................................25 Mice..........................................................................................................25 Transgenic mice...................................................................................25 Mouse lines..........................................................................................26 Histological staining procedures ..............................................................26 In situ hybridization.............................................................................26 Immunofluorescence............................................................................27 Quantification of stainings...................................................................28 Ventral root electrophysiology.................................................................28 Stretch reflex measurements................................................................29 Fictive locomotion ...............................................................................30 Behavioral testing.....................................................................................30 Rotarod ................................................................................................31 Beam Walking .....................................................................................31 Hanging wire .......................................................................................31 Grip strength ........................................................................................32 Digigait ................................................................................................32 Result summary ............................................................................................33 Paper I ......................................................................................................33 Paper II .....................................................................................................34 Paper III....................................................................................................34 Paper IV ...................................................................................................35 Discussion .....................................................................................................37 Novel markers for subtypes of motor neurons and premotor interneurons ..................................................................................................................37 Fast and slow motor neurons ...............................................................38 Gamma motor neurons ........................................................................39 Renshaw cells ......................................................................................40 Partition cells .......................................................................................40 Excitotoxicity and motor neuron subtypes in ALS.....................................40 The influence of 5-ht1d and VIAAT-mediated Renshaw cell signaling on the development of spinal motor circuits .................................................42 Development of the stretch reflex in 5-ht1d-/- knockout mice .............42 Development of spinal motor circuits in the absence of VIAATmediated Renshaw cell signaling.........................................................44 Motor behavior in 5-ht1d-/- and Chrna2::Cre;Viaatlx/lx mice ...................46 The stretch reflex amplitude in locomotion.........................................46 Recurrent inhibition in locomotion......................................................47 Acknowledgements.......................................................................................49 References.....................................................................................................51 Abbreviations 5-HT 5-ht1d AHP ALS Calca ChAT Chodl Chrna2 CNS CPG DRG Egr3 En1 EPSP/EPSC ERR FF FR GPCR GTO IPSP/IPSC LMC MMC NMJ Pitx2 S SMA TrkC VAChT Serotonin Serotonin receptor 1d After hyperpolarization Amyotrophic lateral sclerosis Calcitonin/Calcitonin-related polypeptide alpha Choline acetyltransferase Chondrolectin Cholinergic receptor alpha 2 Central nervous system Central pattern generator Dorsal root ganglia Early growth response 3 Engrailed 1 Excitatory postsynaptic potential/current Estrogen-related receptor beta Fast fatigable motor unit Fatigue-resistant motor unit G protein-coupled receptor Golgi tendon organ Inhibitory postsynaptic potential/current Lateral motor column Medial motor column Neuromuscular junction Paired-like homeodomain transcription factor 2 Slow motor unit Spinal muscular atrophy Neurotrophic tyrosine receptor kinase C . Vesicular acetylcholine transporter Introduction Movement is central for the life of animals. From the relatively simple but vital breathing movements to the subtler act of communicating an emotion through facial expression, in essence what is happening is a movement - a muscle contraction. Most of us perform these behaviors every day without any apparent effort, and they may seem plain, but in fact, correct movement is a delicate interplay between the brain, muscles and the environment. Consider for example the routine act picking up a coffee cup. What you do is to stretch out your arm in a straight trajectory, grab the ear of the coffee cup and lift it to your mouth to have a sip. Consider that movement again: moving your arm in a straight trajectory requires precise activation of just the right ones of the approximately 50 muscles in your upper arm in the correct sequence. Depending on the starting position of your arm and the location of the cup, different sets muscles are activated, still performing the same straight trajectory. Grabbing and lifting the coffee cup to your mouth involves activating even more muscles of the arm and digits, again in the right sequence with just the right force. When the cup reaches your mouth, you need to form a tight seal with your lips around the cup and then activate muscles in your mouth and throat to swallow the coffee. In view of the complexity of this simple act, it is not surprising that most parts of the brain and much of the brains computational power in fact are involved in creating and tuning movements The structures in the central nervous system (CNS) directly involved in movements are the motor cortex, basal ganglia, cerebellum, parts of the brain stem and spinal cord. Lesions to any of these structures in humans or experimental animals leads to deficiencies in movement, from paralysis after a complete transection of the spinal cord to the tremor and difficulties of initiating movements in a patient with Parkinson’s disease caused by a lesion of the substantia nigra of the basal ganglia. Correct and automatic movements also require the integration of information from sensory systems and other parts of the brain. Consider that cup of coffee again. We use visual information to determine the distance to the cup and to estimate the force we need to apply to lift it. When grabbing it we use tactile information to correct for the actual weight. Maybe the cup is unexpectedly hot! That will activate yet another motor program that instead of lifting the cup causes withdrawal of the arm. This type of sensory information is processed in other parts of the brain such as the visual cortices and thalamus, and from these brain regions, 9 the information is fed to the structures regulating movement. When considering such indirect input as a part of the neural correlates of movement it can be proposed that the great majority of the brain is involved in shaping movements. For this thesis on neural control of movement, focus is on the structures in this schema that are in direct contact with muscles. These are the motor neurons, which activate muscles, and proprioceptive sensory neurons, which provide the CNS with feedback on muscle activity. 10 Background Neural control of movement Before going in to the details of the spinal circuits that are the focus of this thesis, the principles and circuits that underlie movement in animals will be briefly introduced to put the function of spinal circuits into context. Numerous neural circuits in the brain are required to generate the full repertoire of movements present in an individual (Figure 1). This neural correlate of movement is organized in a hierarchal system where each level contributes with information necessary for a specific movement. This has been reviewed extensively and is only summarized here [1-3]. The foundation of neural control of movement is formed by the motor neurons. Motor neurons are the only cells in the CNS capable of directly activating muscles. Thus, the motor neurons forms them the final common path of neural activity, as proposed by the English neuroscientist Sir Charles Sherrington [4]. To generate movement, motor neurons integrate information from the supraspinal and sensory structures and transform it into a precise temporal and magnitudal activation of muscles. In direct contact with motor neurons are ensembles of neurons, termed central pattern generators (CPGs), which are capable of generating a rhythmic and patterned output from a tonic input. The pattern provides the framework for motor neuron activation that coordinates muscles for stereotypic movements such as breathing, chewing and swimming. Each stereotypic movement pattern is generated by a specific CPG that is located in the brain stem or spinal cord. Of particular interest for this thesis is the CPG that creates the pattern for hindlimb activation during locomotion. This CPG is located in the ventral spinal cord at lumbar levels. Although the activity generated by CPGs sets the basic rules for motor neuron activation during movements, to adapt to a changing environment this activity must be modulated. Sensory structures and descending pathways from the brain stem are therefore in direct and indirect contact with motor neurons and CPGs to modulate its activity. The modulation entails both correcting for external perturbations, setting the speed and steering and well as initiation of CPG activity. 11 Figure 1. Schematic illustration of a cross-section of the rodent brain. Shaded areas indicate regions involved in motor control. In direct contact with the spinal cord is also the motor cortex. This structure is involved in the conscious control of movement. The direct connection to the spinal cord is used to directly modulate movements. The motor cortex is also involved in the planning of movements. To accomplish these functions, inputs from most areas of the brain converge in the motor cortex and the motor cortex in turn project to many brain areas. Two such areas are the basal ganglia and cerebellum. Basal ganglia is involved in the selection of motor programs by balancing a permissive and inhibitory effect on motor programs. When this balance shifts toward increased inhibitory influence, it is manifested as trouble initiating movements in a Parkinson’s patient. In contrast, when the balance is shifted toward permissive function, it can be illustrated by the constant involuntary movements of a patient suffering from Huntington’s disease. Both are neurodegenerative diseases that affect different parts of the basal ganglia. Sensory information from all senses are integrated in the cerebellum which creates an output that modulates ongoing activity to control the timing, duration and amplitude of movements. Thus, the neural basis of movement is a flow of motor and sensory information that is processed at several levels in many structures at the same time. However, all this information must inevitably end up on motor neurons to generate an effect on the motor behavior of the organism. The motor neuron Several cell types in the brain are called motor neurons. These include the upper motor neurons that transmit motor information within the brain and the preganglionic motor neurons of the autonomic nervous system. However, for this thesis a motor neuron is defined as a neuron with the cell body in the central nervous system that projects its axon to skeletal muscles. This somatic motor neuron is a classical experimental model in neuroscience. It was from motor neurons that the first demonstration of chemical neurotransmission was made [5] and that neurotransmitter release is quantal and vesicular 12 [6]. It was also by studying sensorimotor synapses the molecular principles of memory storage was described, and it was by recording from motor neurons that Sir John Eccles could propose the existence of the axon initial segment and demonstrate the first chemical synapse in the central nervous system on the Renshaw cell [4, 7]. A motor neuron innervates dozens to hundreds of muscle fibers in one muscle. The motor neuron and all the muscle fibers it innervates is called a motor unit. A train of action potentials in a motor neuron causes release of acetylcholine at all muscle fibers it innervates with high fidelity. The motor neuron synapse on the muscle fiber is called the neuromuscular junction (NMJ). Release of acetylcholine at the NMJ activates nicotinic receptors on the muscle fiber, which will start a cascade of signaling events that leads to the contraction of the muscle fiber. This is the essential function of the motor neuron: to evoke muscle fiber contraction. Figure 2. Schematic illustration of the organization of motor neurons in the spinal cord. (A) A spinal cord with cervical, thoracic, lumbar and sacral parts marked. MMC motor neurons are located at all levels of the spinal cord, LMC motor neurons are located only at cervical and lumbar levels. Motor pools are subgroups of the MMC and LMC that innervate one muscle (B) Organization of motor columns in the spinal cord at cervical, thoracic and lumbar levels. Stippled line in (A) denotes level of cross section (C) LMC motor neurons innervate muscles in the limb (left) and MMC motor neurons innervate muscles in the trunk (right). (D) Within the motor columns, individual motor pools innervate different muscles. 13 Organization of motor neurons Motor neurons are located along the entire length of the spinal cord and in discrete nuclei in the brain stem (Figure 2). The spinal motor neurons innervate the muscles of the body while the brain stem motor neurons innervate the muscles of the eye, face, neck, mouth and throat. In the spinal cord, motor neurons are organized in columns based on the location of their target muscles. Motor neurons in the lateral motor column (LMC) innervate the limbs and motor neurons in the median motor column (MMC) innervate the trunk. The LMC innervating the arms or forelimbs are located on cervical levels, while the LMC innervating legs or hindlimbs are located on lumbar levels and the MMC is present at most levels of the spinal cord. Within each column, the motor neurons innervating one specific muscle are clustered in motor pools. The motor pool forms a cigar-shaped entity that spans a couple of segments of the spinal cord. One motor pool contains 20300 motor neurons depending on the muscle [8]. Motor neuron subtypes Within each motor pool, three subtypes of motor neurons exist: alpha, beta and gamma. They are classified based on which type of muscle fiber they innervate (Figure 3). Alpha motor neurons are the archetypal motor neurons. They innervate the force-generating muscle fibers and their activity leads to contraction of muscles. In contrast, activity in gamma motor neurons causes no change in the total length of the muscle. Instead, gamma motor neurons innervate a specialized type of muscle fiber called the muscle spindle, which serves as a muscle length sensor. Beta motor neurons form synapses on both force generating muscle fibers and muscle spindle fiber. Figure 3. Schematic illustration of subtypes of motor neurons. Alpha motor neuron innervating extrafusal force-generating muscle fiber, gamma motor neuron innervating intrafusal muscle spindle fiber and beta motor neuron innervating both extra- and intrafusal muscle fiber. 14 Alpha, beta and gamma motor neurons lie intermingled with each other in the motor pool [9]. Morphologically, both alpha and gamma motor neurons have large dendritic trees [10]. However, looking at the soma area, gamma motor neurons are smaller than alpha motor neurons and have a thinner axon, which is reflected in a slower conduction velocity. Alpha and gamma motor neurons also receive different synaptic inputs. Alphas receive socalled C-boutons and M-boutons contacting their soma, while gammas do not [11, 12]. C-boutons are cholinergic terminals derived from partition cells (see discussion) and M-boutons are from Ia sensory neurons [13, 14]. Slow and fast motor units Alpha motor neurons can be further subdivided based on the type of muscle fiber it innervates. Mammalian muscles are composed of two types of muscle fiber: slow-twitch and fast-twitch. The basis for this classification is the duration of the twitch contraction time [15]. Slow fibers (S) also generate a smaller force and are more resistant to fatigue than fast fibers. Fast motor neurons can be further subdivided into fatigable (FF) and fatigue-resistant (FR) based on the duration they can sustain contraction. Most mammalian muscles consist of a mix of slow and fast fibers in different ratios depending on how the muscle is used. This can be illustrated by two muscles in the calves: gastrocnemicus and soleus. Gastrocnemicus, which is used for example for jumping, has a low slow/fast ratio while soleus, which is used for postural control, has a higher slow/fast ratio [16]. The motor neurons innervating slow and fast muscle fibers exhibit discrete properties. This was first shown by Eccles et al for afterhyperpolarization (AHP) duration [17]. AHP is the phenomena where the membrane potential after an action potential undershoots the resting membrane potential. Slow motor neurons have a longer after-hyperpolarization time than fast motor neurons. The functional consequence of this is that slow motor neurons have a longer “waiting period” between action potentials and thus cannot fire in the same frequency as fast motor neurons. Subsequent recordings from both cat and rat have shown that fast and slow motor neurons differ in other electrical properties [18, 19]. These include input resistance, a measure of the resistance over the plasma membrane, and rheobase, a measure of the current needed to generate an action potential. Slow motor neurons have a higher input resistance, which is one of the underpinnings of Henneman’s size principle of motor unit recruitment. Henneman’s size principle dictates that slow (smaller) motor units will be recruited first when activating a muscle, after that (larger) fast fatigue-resistant and then fast fatigable motor units will follow [20, 21]. Thus, a slow movement generating smaller force will include only slow motor units, while increasing the speed or force of the movement will include fast motor units as well. 15 Development of motor neurons The CNS contains a great diversity of different cell types. Just in the spinal cord, it is estimated that hundreds of different cell types are formed during development, each with a different character [22]. As has already been outlined, motor neurons are categorized as alpha, beta and gamma. Alphas exist as slow, fast fatigue-resistant and fast fatigable, and gammas exist as dynamic and static (description of subtypes of gamma motor neurons will follow in next section). In addition, motor neurons are located in different columns and in motor pools, each with a specific target muscle. Neural diversity is created in the spinal cord by the generation of distinct progenitor domains in the dorsoventral axis. The timing and concentration of morphogen factors such as sonic hedgehog, fibroblast growth factors and retinoic acid induces the expression of transcription factors that acts mutually exclusive to define specific progenitor domains [23-25]. The progenitor domain that generates all motor neurons is designated pMN and is defined by the expression of basic helix-loop-helix protein Olig2 and the homeodomain transcription factors Pax6, Nkx6.1 and Nkx6.2 [26, 27]. When progenitor cells in the pMN exit the cell cycle, they start expressing the homeodomain transcription factors Hb9, Isl1 and Isl2 that defines the generic motor neuron fate [28-30]. The development of columnar and motor pool identity is acquired in a similar way where a extracellular morphogen gradient is translated into a code of in this case Hox transcription factors that act mutually exclusively to define boundaries for the motor columns and motor pools in a process that requires FoxP1 [31-33]. Encoded in these transcription factor networks is the information on where to settle in the spinal cord, what neurotransmitter to use and how to project the axon in the periphery. In contrast to the well-defined extrinsic and intrinsic signals that are directing the subtype-specification of motor neurons into columns and pool the molecular mechanisms that specifies alpha, beta and gamma motor neurons have not been described. In the adult spinal cord, gamma motor neurons express Hb9 and Isl1, but if that is also true in the embryonic spinal cord is not known [34]. However, gamma motor neurons appear to develop later in embryogenesis than alpha motor neurons as gamma axons reach the muscle spindles as late as E18.5 in the extensor digitorum longus muscle while alpha motor axons form NMJs at E14.5 [35]. In addition, small putative gamma motor neurons are generated later in development than large putative alpha motor neurons [36]. Similar to for alpha, beta and gamma motor neuron character, the slow and fast character of alpha motor neurons has not been described molecularly in the embryo. However, extensive studies on chick embryos has shown that when given the choice, fast motor axons preferentially innervate fast motor fibers and vice versa, suggesting that fast and slow motor neurons develop properties specific for its subtypes to find and synapse with the cor16 responding muscle fiber in the embryo [37-40]. It should be noted though that although fast and slow motor neurons likely are specified early in spinal cord development, the adult fast and slow motor units are plastic. Motor units can be converted from a fast to slow and vice versa by alterations in the firing frequency of the innervating motor neuron [41, 42]. Thus, the fast and slow motor neuron character of a motor neuron appear to be specified genetically in the embryo, but can adapt its character to match requirements of changed activity in the adult. Degeneration of motor neurons in ALS Neurodegenerative diseases are characterized by a progressive loss of neural function. Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease that affects the motor neurons. People suffering from ALS typically experience muscle weakness that progresses over time and eventually lead to complete paralysis. Although most cases of ALS cannot be attributed to genetic traits, 5% of the cases are inherited. Of these, 20% have a mutation in the enzyme superoxide dismutase 1 (Sod1) [43]. Clinically, both sporadic and inherited ALS has indistinguishable characteristics and mice expressing mutant Sod1 develop a disease similar to human ALS [44]. In ALS, both the upper and lower motor neurons degenerate by unknown mechanisms. A number of pathologies have been identified in both humans and mouse models of the disease including protein aggregates, excitotoxicity and deficiencies in retrograde transport [44]. However, it is not established whether these pathologies are causing disease or are the result of disease. Of particular interest for this thesis is the excitotoxicity observed in motor neurons during disease. Excitotoxicity is caused by an excessive glutamatereceptor activation leading to a massive Ca2+ influx that will cause damage to the mitochondria and eventually apoptosis. In ALS patients and the Sod1G85A mouse model of ALS, diminished glutamate transport has been reported as well as a reduced expression of the glutamate transporter EAAT2 in the motor cortex and spinal cord [45, 46]. In addition, mice heterozygous for EAAT-2 expression develop disease earlier than littermates while drugs that enhance EAAT2 activity prolong life span [47-49]. Another feature of degeneration of motor neurons in ALS of interest for this thesis is the selective degeneration of subtypes of motor neurons. In ALS patients as well as mouse models of ALS, the degeneration of motor axons is predictable and follows a scheme of decay [50, 51]. In this scheme, FF motor units degenerate first at P50, FR follows at P80 and S units remain until death [51]. Thus, selective vulnerability of motor neuron subtypes appears to be present in ALS. 17 Proprioception We constantly have a sense of where our limbs and body are located in relation to each other in the three-dimensional space. Even with our eyes closed, and even those blind from birth, can tell if their arm hanging along the side of the body or extended into the air. The sense that makes this possible is called proprioception. Unlike the classical senses like vision and olfaction that transmits signals about the state of the external world to the brain, proprioceptive sensors reports how the limbs, trunk and head are positioned and moving in relation to each other. Proprioception is not necessary for movement, but is required for a smooth and automatic movement. This can be illustrated by the story of Ian Waterman, who suffered an unusual degeneration of proprioceptive sensory neurons [52]. The degeneration, likely caused by a viral infection, led to a specific loss of proprioceptive signaling below the neck while sparing other somatosensory modalities such as pain, temperature and deep pressure. Just after the loss of proprioception, Waterman was unable to maintain his posture or to initiate voluntary movements suggesting that proprioceptive information is an integral part of the neural activity that generates an upright position. Through practice, he would learn to move and eventually walk again, although his gait lacks the fluidity and dynamics of normal gait [53]. The most striking thing with his newly developed gait though, is that every step is a conscious act that requires constant visual feedback for him to control his limbs. Proprioception is dependent on information from joints, skin and muscles. The modalities signaled by these structures include joint extension/flexion, skin stretch, muscle stretch and muscle force [54, 55]. This information is signaled from specialized structures in the target organs to the spinal cord where it is integrated in spinal circuits that relay the signal to the brain or create a motor output. For this thesis, the proprioceptive information on muscle stretch is under focus. This information comes from the muscle spindles. Anatomy and physiology of the muscle spindle The muscle spindle is a fusiform encapsulated organ that is embedded within muscles [56]. It is composed of a specialized type of intrafusal muscle fiber. Muscle spindles are positioned in parallel with the force-generating extrafusal muscle fibers. They are predominantly found within the deep belly region of the muscle, where slow muscle fibers predominate. In cats, the number of spindles per muscle range from 10 in a small muscle to over 100 in a large muscle and the total amount in all human muscles is estimated to be 25 000 muscle spindles [57, 58]. 18 Figure 4. Schematic illustration of muscle spindle innervation. Ia afferents forms annular terminations in the central domain. Group II afferents form “flower-spray” terminations adjacent to Ia afferent and gamma and beta motor neurons forms synapses at the polar ends. When a muscle is stretched, the muscle spindle is also stretched. This activates sensory neurons in contact with the muscle spindle. Information from the muscle spindle is transmitted to the spinal cord by two types of sensory neurons: group Ia and group II. Group Ia afferents form annular terminations around the central domain of the muscle spindle, while group afferents form so-called flower spray terminations (Figure 4). Group Ia and II afferents signal different overlapping modalities of muscle stretch. When a passive muscle at length x is stretched to a new length y, Group Ia afferents fire action potentials as the muscle is being stretched. The frequency of this signal is dependent on the velocity of the stretch. Thus, Ia afferents provide the CNS with information on the velocity of length change. Group II afferents on the other hand signals at one frequency at length x and at a higher frequency at length y. Thus group II afferents will provide the CNS with information on how stretched a muscle is. As previously mentioned, muscle spindles also receive motor innervation from beta and gamma motor neurons. A signal from a gamma or beta motor neuron contracts the muscle spindle fiber. This contraction is so small that it does not generate a contraction of the whole muscle, but merely lead to contraction of the muscle spindle. This contraction leads to increased sensitivity of group Ia and II sensory afferents. Subtypes of gamma motor neuron have been described that relate to the modality of muscle spindle stretch sensitivity they regulate. Dynamic gamma motor neurons increase the sensitivity of primarily Ia afferents to dynamic changes in muscle spindle length and static gamma motor neurons increase the sensitivity of primarily group II afferents [59]. This motor innervation provides a system for the CNS to adjust proprioceptive feedback depending on the task. For example walking on level ground requires less sensitive proprioceptive feedback than balancing on a plank. Thus, gamma motor neurons are more active in the balancing act than when walking on the floor. The motor innervation to muscle spindles also makes sure that muscle spindles move in concert with extrafusal fibers. If a muscle would contract while the muscle spindle don’t, then it would slacken and group Ia and II sensory neurons wouldn’t signal during muscle contraction. Therefore, alpha 19 and gamma motor neurons in one motor pool are often active at the same time in the locomotor cycle, which is referred to as alpha-gamma linkage. Proprioceptive circuits The sensory signals are transmitted to the spinal cord where they are integrated into spinal circuits to modulate ongoing movement and relayed to the brain. In the spinal cord, group Ia afferent makes contacts with Ia inhibitory interneurons, dorsal spinocerebellar tract neurons and alpha motor neurons. Group II afferents contact group II interneurons in the intermediate and deep dorsal horn and presumably gamma motor neurons (Figure 5) [60-62]. The connection between alpha motor neurons and Ia afferents provides alpha motor neurons in one motor pool with monosynaptic feedback from muscle spindles in the same muscle it innervates. This circuit is activated when testing the knee jerk reflex (also called stretch reflex). Ia afferent information from that same muscle signal to Ia inhibitory interneurons that inhibit motor neurons innervating antagonist muscles. The third central connection of Ia afferents is to spinocerebellar projection neurons. The spinocerebellar neurons relay proprioceptive signals to the cerebellum. The group II interneurons are a loosely defined group of interneurons that innervate alpha and gamma motor neurons and other interneurons in the spinal cord [63, 64]. The role of the group II interneurons is not well defined, but given their monosynaptic input from group II afferents about the length of muscles and close contact with motor neurons they likely have a role in coordinating muscle activity [64]. Figure 5. Schematic illustration of central terminations in the spinal cord formed by muscle spindle afferents in the lumbar spinal cord. Group Ia afferents terminate dorsal to the central canal on Clarke’s column neurons, in the intermediate horn of Ia inhibitory interneurons and on alpha motor neurons. Group II afferents terminate in the deep dorsal horn on group II interneurons and possibly in the ventral horn on gamma motor neurons. 20 Group Ia and II afferents are both subject to presynaptic inhibition from local interneurons in the spinal cord as well as descending tracts [65, 66]. Presynaptic inhibition is a mechanism to centrally inhibit sensory signaling when it is required. Presynaptic inhibition can also serve to change the relative importance of a certain proprioceptive synapse, e.g. by silencing the synapse between group Ia-alpha motor neurons contacts while Ia-Ia interneuron contacts signal at normal rates. Development of the monosynaptic stretch reflex Similar to the development of motor neurons, sensory neurons in the DRG are specified by the action of morphogens that activate transcriptional programs unique for each sensory neuron subtype [67]. Upon acquiring its character, proprioceptive sensory neurons can be identified by the expression of the RUNT-domain transcription factor Runx3 and the neurotrophic tyrosine receptor kinase-C (TrkC). Runx3 is required for proprioceptive neuron survival and acts in a concentration-dependent manner to determine the laminar termination of sensory axons in the spinal cord [68, 69]. Proprioceptive neurons follow motor axons out to the muscle [70]. The proprioceptive neurons require the motor neurons, as ablation of motor neurons leads to an absence of muscle innervation of sensory neurons [71]. When entering the muscle, the Ia afferent neuron induces the formation of a muscle spindle from primary myocytes by signaling through neuregulin 1 [72]. This activates expression of the zinc-finger transcription factor Egr3, and ETS transcription factors Er81 and Pea3 that serve to differentiate the muscle spindle [72-74]. Figure 6. Activity-dependent and retrograde influence on development of the stretch reflex. Reducing the stretch reflex activity leads to increased synaptic strength in the Ia-motor neurons synapse (left). Removing Nt-3 production from the muscle spindle lead to reduced synaptic strength in the Ia-motor neurons synapse (right). In the spinal cord, Ia afferents project with high precision to the correct motor pool in an activity-independent manner using genetically specified axon guidance molecules [75-77]. Maturation of the circuit, on the other hand, forms in an activity-dependent manner involving target-derived molecules 21 from the muscle spindle (Figure 6). Reducing stretch reflex activity by tetrodotoxin application to the nerve, d-tubocurare treatment or axotomy lead to increased synaptic input from Ia afferents to motor neurons [76, 78, 79]. In muscle-specific ErbB2 knockout and Egr3 knockouts where muscle spindle develop abnormally, appropriate connections form with the correct motor pool but the dorsal root-evoked EPSP is decreased in amplitude and the number of proprioceptive synapses are reduced [80]. Muscle spindle-specific deletion of Nt-3 also leads to reduced reflex amplitude, however it is delayed compared to spindle-specific ErbB2 knockouts [80]. Thus, Nt-3 is one of the muscle spindle-derived factors that regulate the amplitude of the monosynaptic stretch reflex, but likely, other factors are involved as well. Recurrent inhibition Birdsey Renshaw observed that upon antidromic stimulation of the ventral root, excitation of motor neurons by Ia afferents were reduced [81, 82]. John Eccles and colleagues showed that this effect was caused by cholinergic activation of an inhibitory cell type in the ventral horn, which he named the Renshaw cell in honor of its discoverer [83]. Figure 7. Schematic illustration of the recurrent inhibition circuit. RC, Renshaw cell. MN, motor neuron. The Renshaw cell-Motor neuron circuit is as simple as it is vexing (Figure 7). Concomitantly with sending action potentials to the muscle, alpha motor neurons signal via an axon collateral to Renshaw cells in the spinal cord. This activates the Renshaw cell, which respond with a high-frequency burst that inhibits the same motor neuron. The location of the Renshaw cell close to the motor neuron, “eavesdropping” on all motor neuron activity, puts it at a central position for motor output. Still, there is no conclusive data on what function the Renshaw cell has in movement. 22 The motor neuron uses acetylcholine and another unknown neurotransmitter that activates NMDA receptors to signal to Renshaw cells [84-86]. This generates a high-frequency burst of Renshaw cells that in turn use glycine and GABA to signal to motor neurons [87]. Only alpha motor neurons activate Renshaw cells, as gamma motor neurons do not have intraspinal axon collaterals [10]. Different motor unit types activate Renshaw cells with different intensity, where FF motor neurons generate a larger recurrent IPSP than S motor neurons, however, the response to this IPSP is equally large in all motor unit types [88]. Development of recurrent inhibition Renshaw cells develop from the progenitor (P)1 progenitor domain [89]. As P1 neurons mature into ventral (V)1 neuron they start expressing the homeodomain transcription factor engrailed 1 (En1) and send axons to motor neurons in a guidance process involving Netrin [90]. The functional development of Renshaw cells has mainly been studied in chick embryos where Renshaw cells are called R-interneuron [91]. These studies have shown that the first axonal connections is likely formed by Rinterneurons on motor neurons and later in development motor neurons send axonal collaterals to innervate R-interneurons [92]. The connection between R-interneurons and motor neurons has been proposed as the source of episodes of rhythmic spontaneous activity [93, 94]. In the embryonic nervous system, such spontaneous activity in developing circuits is believed to shape the connectivity, synaptic strength and excitability as the circuits mature [95]. This has been observed in the chick spinal cord where blocking spontaneous activity leads to changes in excitability and synaptic strength [96-98]. In the mouse spinal cord, where this spontaneous activity starts around E12.5 and is mediated by acetylcholine, glycine and GABA [99] disruption of spontaneous activity by knockout of ChAT leads to abnormal development of spinal motor circuits and motor axons makes guidance errors in the developing limb [100, 101]. In addition to the suggested role in spontaneous episodes of rhythmic activity in the spinal cord, Renshaw cells have transient features during development. GABA and glycine are excitatory during development, which means Renshaw cells will exert recurrent facilitation on motor neurons [92]. In addition, Renshaw cells receive synaptic contacts from Ia afferents during development [102]. The excitatory action of Renshaw cells reverses as chloride homeostasis change in the spinal cord (E15.5 in mouse) and the Ia afferent input to Renshaw cells wane [102, 103]. Thus, it is possible that Renshaw cells have a specific role during development. 23 Aims The aims of the work in this thesis are: To find and characterize novel molecular markers expressed in subpopulations of neurons involved in locomotion To determine how expression of the fast and slow motor neuron markers Chodl, Calca and ERR are affected in the Sod1G93A mouse model of ALS To determine the role of 5-ht1d in development of proprioceptive circuits To determine the contribution of VIAAT-mediated Renshaw cell signaling to development of spinal locomotor circuits 24 Methodological considerations This section provides a short discussion of the principles and validity of the methods used in the experiments for this thesis. Detailed information of the procedures can be found in the respective paper. Only methods performed by the author of this thesis are presented. Mice The experiments presented in this thesis were conducted on mice. All procedures were approved by the local Swedish ethical committee (permit C147/7 and C79/9) or by the University of Calgary Animal Care Committee. Mice were chosen as the model system because they are available for transgenic use and they are an established model system for locomotion with welldescribed histological, electrophysiological and behavioral protocols. Although other species are equally well suited for transgenic use, such as Drosophila melanogaster, C. elegans and zebrafish, these systems are not as developed for studies of motor behavior. It can be argued that the rat is the model organism of choice for motor behavior, however it was only until recently that rats were available for transgenic use [104]. Therefore, to study neural circuit development and function and to relate this information to motor behavior the mouse was chosen as the model organism. Transgenic mice Transgenic animals have been used in all papers included in this thesis. The genome of a transgenic mouse has been modified by genetic engineering. Such modification can be the deletion of a gene (knockout) or insertion of a foreign gene such as Cre to target floxed alleles or mutant Sod1 to create a disease model. Knockout animals are valuable tools that can be used to study gene function. Removal of a gene of interest and subsequent studies of the cells, circuits and behavior may give information on the function of the gene. However, since most genes are expressed in more than one cell type, a knockout of a gene may affect several cell types other the one being studied. By default, the gene is also knocked out already from conception, which means that eventual compensations for the knockout during embryonic develop25 ment always need to be considered. In these instances, the Cre/lox-system is useful. Cre is a site-specific recombinase that recognizes a 34 bp sequence called loxP. If loxP-sites are introduced flanking a gene of interest and Cre is expressed from a promoter specific for the cell of interest, it will generate a conditional knockout of the gene only in cells where both promoters are active at the same time. In addition to creating cell-specific knockout, the Cre/lox system can also be used to drive expression of reporter molecules such as GFP that facilitates finding a cell type for patch-clamp recordings or to visualize cell bodies and neurites. Mouse lines For in situ hybridizations, immunohistochemistry, retrograde tracings and microarray experiments of wild type tissue, the inbred C57/BL6 line was used. For electrophysiology in paper I Swiss Webster mice was used. The transgenic mouse lines used in the thesis are described in Table 1. Transgenic animals were genotyped using PCR on isolated genomic DNA using the primers described in papers I-IV. Histological staining procedures In situ hybridization In situ hybridization is a technique for labeling nucleic acids by taking advantage of their ability to bind to a complementary strand of nucleic acid [105, 106]. The complementary nucleic acid, called a probe, is labeled with an epitope tag. An antibody coupled to an enzyme binds to the tag and when a substrate for the enzyme is added, the location where the probe/antibody/enzyme-complex has bound is visualized. For the experiments in this thesis, both chromogen and fluorescent substrates have been used interchangeably without any noticeable difference in sensitivity. The fluorescent staining works better to combine with immunohistochemistry. In Paper I, double in situ hybridization was used. In double in situ hybridization, two transcripts can be located on the same tissue sample and stained in different colors. First, an antibody complex binding to one of the probes is developed in one color. Then the enzyme is inactivated and the antibody/enzyme complex recognizing the second probe is developed in another color. If inactivation is incomplete, the first probe will also develop together with the second probe, which means careful controls are required for double in situ hybridization. 26 Table 1. Transgenic mouse lines used in this thesis Name Locus Transgene description Obtained from Reference Targeted mutation of the zinc-finger DNA-binding domain and carboxy terminal of Egr3 Warren Tourtellotte (Northwestern Egr3-/Egr3 University) [74] James F Martin (Texas A&M System Health Science Center) Pitx2-Cre Pitx2 [107] Targeted integration of a Silvia Arber (Frielox-STOP-lox-mGFP-IRES- drich Miescher NLS-LacZ-pA cassette into Institute for BioMapt exon 2 of the Mapt gene. medical Research) [108] TaumGFP-nlslacZ Exons 4, 5 and 6 were Vglut2lx/lx Slc17a6 flanked with loxP sites Generated in our lab [109] A plasmid with Sod1G93A driven by the human Sod1 Jackson labs, Bar [110] Sod1G93A Random promoter Harbour, ME A plasmid with Cre driven by the PGK promoter PGK-Cre Random [111] Targeted mutation of the MMRRC/Lexicon coding exon of Htr1d. genetics 5-ht1d-/Htr1d [112] In a BAC containing the htr1d gene, htr1d was re5-ht1d::GFP Random placed with GFP MMRRC/GENSAT [113] In a BAC containing the Chrna2 gene, Chrna2 was Chrna2::Cre Random replaced with Cre Generated in our lab Slc32a1 Exon 2 of the Slc32a1 gene Bradford B. Lowell [114] Viaatlx/lx was flanked with loxP sites (Harvard Medical School) R26::lsl.Tomato Gt(ROSA)2 A CAG-lox-stop-loxAllen brain institute [115] 6Sor tdTomato-WPRE-pA plasmid was inserted into the Gt(ROSA)26Sor locus Immunofluorescence Immunofluorescence takes advantage of the specific binding of an antibody to its epitope. The technique is useful to determine the localization of proteins in a tissue sample. In this thesis, immunofluorescence has been used to co-localize proteins in synapses and cell body and to visualize axons in the muscle. Immunofluorescence has also been combined with retrograde tracings, in situ hybridization and fluorescent protein expression from transgenic mice. When using antibodies it is important to be careful in the interpretation of stainings [116]. Antibodies are notorious for producing background and unspecific staining. In this thesis, precaution has been made to make sure stain- 27 ing is representative by comparing them to previously reported expression pattern and with matching in situ hybridizations. Quantification of stainings Quantification of stainings in the current thesis has been performed manually after in situ hybridization or immunofluorescence. To make confident estimates of the number of cells expressing a certain gene after in situ hybridization, only cells with a complete staining around the nucleus was counted. When possible, we only counted sections from the same spinal segment (e.g. L1-L3), however some quantification was made from the same column (e.g. lumbar LMC). The number of synapses in contact with motor neurons or Renshaw cells was quantified in Paper III and IV. A synapse was in most stainings defined by the expression of the synaptic protein synaptophysin [117]. If co-staining with synaptophysin was not performed, a synapse was defined as a bulging of a stained axon in close contact with a ChAT-stained motor neuron (e.g. Calbindin and Tomato) or by strong expression of a subtype-specific vesicular transporter (VAChT). To obtain comparable results, the synapses quantified were from the same level of the spinal cord and the confocal images were of the same optical thickness and number. The same criteria were used for the quantification of muscle spindle innervation. In paper I and III, soma area of motor neurons were measured after in situ hybridization with a probe binding VAChT mRNA. Care was taken not to overdevelop the staining as this could lead to an overestimation of soma size. Soma areas were measured by manually drawing the outline of the stained area as illustrated in figure 3L in paper I. Soma area quantifications were performed using either Volocity software (Paper I) or ImageJ software (Paper III). Ventral root electrophysiology With electrophysiology, the flow of ions across the cell membrane that constitute the basis for neural activity is measured. By attaching an electrode to the ventral root, the population activity of motor neurons projecting through that root can be measured. For the experiments in this thesis, the spinal cord was dissected from the animal and kept alive for several hours in artificial cerebrospinal fluid (aCSF). 28 Stretch reflex measurements Figure 8. Schematic illustration of experimental setup for ventral root electrophysiology. (A) For measurement of stretch reflexes a stimulus electrode is attached to the dorsal root and recording electrode to the ventral root. The response is recorded in the ventral root to dorsal root stimulation. (B) For measurement of druginduced fictive locomotion electrodes are attached to left and right L2 and L5 ventral roots. The spinal cord is perfused with oxygenated aCSF containing 5 m NMDA and 10 m 5-HT. This induces a CPG rhythm of left-right and flexion extension coordination. To measure sensorimotor connections, a recording electrode is attached to the ventral root and a stimulus electrode is attached to the dorsal root (Figure 8). Experiments were performed in hemisected spinal cords to ensure oxygenation and to avoid presynaptic inhibition from contralateral sources [118]. A 0.2 ms stimulus pulse of increasing intensity was applied until the amplitude of the response in the ventral root did not increase any more. This was defined as the maximal amplitude. For recordings, a stimulus 1.5x of the lowest stimulus that generated maximal amplitude was applied to ensure that all Ia synapses were activated. Stimulation of the dorsal root with this intensity activates motor neurons and many interneurons in the spinal cord. The recorded response in the ventral root is seen as a short first deflection and a longer second deflection. The first deflection with an onset 6 ms after stimulation, mainly corresponds to a monosynaptic activation. The second deflection corresponds to polysynaptic activation. The response in the ventral root depends on several factors: synaptic strength, number of Ia synapses, number of motor neurons, membrane prop- 29 erties of motor neurons, number of afferents activated (tightness of dorsal electrode) and recording of response (tightness of ventral electrode). Two stimulus protocols have been used. In paper III stimulus pulses were given at 1 Hz and in paper IV stimulus pulses were given at 0.033 Hz. A 0.1 Hz stimulus induces a short-term depression at the Ia-MN synapse, which means that the maximal response will be underestimated and the synaptic response recorded in the ventral root will be affected, by such shortterm depression [119]. Fictive locomotion The ability of the locomotor CPG in the spinal cord to evoke rhythmic activity in the absence of sensory input or descending drive is taken advantage of in this method. Electrodes are attached to the ventral roots of the isolated spinal cord to measure the signals of motor axons (Figure 8). Usually, lumbar level (L)2 and L5 are used. L2 innervates mainly hip flexor muscles and L5 innervates hip and ankle extensor as well as some hip flexor muscles [8, 120]. When serotonin and NMDA are added, the activity in the ventral roots is similar to the activity that is seen during locomotion with an alternation of left-right and flexion-extension [121]. The output from the ventral roots is a rhythmic activity that is coordinated. To analyze the frequency of the rhythm, the phase relationship and the coherence we used continuous wavelet transform. This gives a timefrequency representation of the recorded rhythm that can analyze long stretches of data [122, 123]. Before this type of analysis, the raw data was rectified. Analysis was performed in the Matlab-based software Spinalcore [123]. To analyze individual burst parameters we applied a time-series analysis using a custom-written program in Matlab [124]. To calculate the burst duration data was high-pass filtered at 0.1 Hz, rectified and low-pass filtered at 5 Hz. An algorithm was used to determine the length of time between the increase in ventral root discharge rate to half-maximum and the following decrease to half-maximum. This defined the number of bursts and the burst duration and from this data, interburst duration and cycle period was calculated. Behavioral testing To study behavior is to measure the product of neural activity in the complete organism. To unambiguously relate activity in a specific set of neurons to behavior should provide a good estimate of the function of that set of neurons for the organism. In this thesis, various motor tests have been per30 formed to test how alterations in proprioceptive signaling and recurrent inhibition translate into deficiencies in movements. Rotarod The rotarod was designed to measure neurological deficits in rats and mice [125]. The mouse is placed on a steady rod ( 1¼ ) and then the rod starts rotating. The rotation speed is either constant (Paper IV) or linearly accelerating from 0-45 rpm over 60 seconds (Paper III and paper IV). The latency until the mouse falls down is measured. To stay on the rotating rod the mouse has to coordinate its body and balance itself while compensating for the rotation of the rod. In the accelerating rotarod, the mouse has to compensate for the acceleration as well. Deficiencies in rotarod performance have been observed in mice with connectivity deficiencies of En1+ cell including Renshaw cells and in TrkC mice lacking proprioceptive sensory neurons [126, 127]. Beam Walking The beam walking test was designed to measure motor cortex deficiencies in rats but have later been shown to measure locomotor deficit in mice as well [128, 129]. The mouse is put in one end of a one meter long round wooden stick. The starting point is brightly lit and the other end is darker with and enclosed space containing food pellets. Each mouse was given three trials to complete the task in 15 seconds. If the mouse was not crossing the beam, it was counted as a failed trial. While the mouse runs to the other end of the beam, it is videotaped from behind. The videotaped is analyzed for foot slips of the hindlegs. Data is presented as slips per successful trial. If a mouse didn’t have any successful trials, it was removed from analysis. To be able to cross the beam, the mouse has to maintain its equilibrium, with a narrow base of support. In addition, correct foot placement is constrained to the narrow beam. Impaired behavior in beam walking have been observed in rats treated with pyridoxine, which cause degeneration of proprioceptive sensory neurons [130]. Hanging wire Hanging wire is a test of motor strength. The mouse is put on a horizontal thin steel wire with its forepaws and is carefully released by the tail. Scoring is made by judging if the mouse manages to pull itself up with at least one of its hind paws onto the steel wire directly ( 1 second) of if it struggles ( 1 second). Each mouse is tested three times and the best trial is used for analysis. To accomplish this task the mouse has to pull its own weight and coordinate its body to grab the steel wire with the hindpaws. 31 Grip strength To measure maximal grip strength the mouse grips a wire mesh with its forepaws and is pulled back horizontally by a steady slow movement. The force detector records the force when the mouse releases the grip. Each mouse was tested 5 times and the average value was used for analysis. Digigait The Digigait system (Mouse specifics) was used to analyze gait [131]. Each animal was allowed to run on a treadmill with a clear belt while being videotaped from underneath with a high-speed camera. 1000 frames (10 seconds) of video were analyzed using the Treadscan software (Mouse specifics) to detect stance and swing phase for all limbs in each frame. The software determines the time each paw is placed on the belt and from this extracts basic gait parameters such as stride length, swing and stance phases, and stepping frequency. 32 Result summary Paper I Paper I describes a screen for genes expressed in subpopulations of cells in the ventral spinal cord. By comparing gene expression in the ventral compared to the dorsal lumbar spinal cord using a microarray hybridization approach and by selecting interesting candidates from the digital gene expression atlas Genepaint, a shortlist of 341 candidate genes was generated. The specific expression of these genes was analyzed using in situ hybridization. The in situ hybridizations revealed that among the 341 genes, 57 were expressed in a pattern reminiscent of cholinergic neurons (including motor neurons) and 102 were expressed in a pattern suggesting expression in both motor neurons and non-cholinergic interneurons. Three of these genes, Chondrolectin (Chodl), Calcitonin/Calcitoninrelated polypeptide alpha (Calca) and estrogen-related receptor beta (ERR ) were expressed in a pattern suggesting they were specific to subpopulations of motor neurons and one gene, paired-like homeodomain transcription factor (Pitx2) in cholinergic interneurons. All these genes were co-expressed with the vesicular acetylcholine transporter (VAChT), which suggest that they use acetylcholine as their neurotransmitter. Double in situ hybridizations revealed that Calca and Chodl were expressed in the same motor neurons whereas ERR was expressed in a complementary population of motor neurons. The Calca+/Chodl+ motor neurons had a larger soma area than the ERR + motor neurons and whole-cell patch clamp-recordings revealed that Chodl+ motor neurons had a shorter afterhyperpolarization half-decay and a larger rheobase current. These data suggest that the Calca+/Chodl+ motor neurons represent a population of fast motor neurons. The complementary expression pattern of ERR in small motor neurons taken together with an observation that ERR -expression remains in early growth factor 3 (Egr3) mutant mouse which lacks gamma motor neurons suggest that ERR is expressed by slow motor neurons. Expression of Chodl started in the motor neuron region at E10.5 and ERR was expressed from E11.5. Pitx2 was expressed in cholinergic interneurons close to the central canal. Expression started at E14.5. At this stage only 20% Pitx2 was expressed in cholinergic cells. The remaining non-cholinergic cells did not express vesicular inhibitory amino acid transporter (Viaat) and therefore wasn’t inhibi33 tory either. From P11 however, all Pitx2 cells in the lumbar spinal cord were cholinergic. Genetic tracing of Pitx2-cells using a Pitx2-Cre mouse revealed that the Pitx2 cells formed cholinergic synapses on motor neurons suggesting they could be the origin of the C-bouton. Paper II In paper II, the Sod1G93A mouse model of ALS was crossed with mice heterozygous for vesicular glutamate transporter 2 (Vglut2) to generate Sod1G93A;Vglut2+/- mice. In Sod1G93A;Vglut2+/- mice, VGLUT2-expression is reduced and presumably, excitotoxic load on motor neurons is reduced as well. In Sod1G93A;Vglut2+/- mice, both survival and disease onset followed a similar time-course as Sod1G93A;Vglut2+/+. However, motor neuron survival was increased in the Sod1G93A;Vglut2+/- mice measured after in situ hybridization and immunofluorescence. Tibialis anterior also contained more innervated NMJs in Sod1G93A;Vglut2+/- mice. In the brainstem, neurodegeneration of the facial nucleus was rescued by the Vglut2 mutation whereas motor neurons in the hypoglossus nucleus were unaffected in both Sod1G93A;Vglut2+/- and Sod1G93A;Vglut2+/+. Analysis for the expression of the markers for fast and slow motor neurons described in paper I was also performed. Expression of Chodl was completely abolished in Sod1G93A;Vglut2+/- and Sod1G93A;Vglut2+/+ whereas expression of Calca and ERR was reduced but not absent in both mutants. Paper III In parallel with the screen described in paper I, another database search was performed using the GENSAT database. This yielded another marker for a subpopulation of motor neurons, the serotonin receptor 1d (5-ht1d). 5-ht1d was co-expressed with VAChT and soma area estimation showed that 5-ht1d was expressed in the smallest motor neurons. In early growth response 3 (Egr3) knockout mice, the number of motor neurons expressing 5-ht1d was reduced by 90%. These data suggests that 5-ht1d is expressed by gamma motor neurons. In addition to this, 5-ht1d was expressed in proprioceptive sensory neurons. To characterize the electrical properties of gamma motor neurons wholecell patch clamp recordings was conducted on motor neurons in the 5ht1d::GFP transgenic mouse. No differences were seen between 5ht1d::GFP+ (gamma) and 5-ht1d::GFP- (alpha) motor neurons. However, when the alpha motor neurons were subdivided into fast and slow motor 34 neurons using hierarchal clustering method gamma motor neurons could be clustered as a group. To study the contribution of 5-ht1d to development of proprioceptive circuits 5-ht1d-/- knockout mice was analyzed. In 5-ht1d-/- mice, gamma motor neurons survived and muscle spindles showed normal innervation by both gamma motor neurons and sensory neurons. In 5-ht1d-/- mutants, the monosynaptic activation of motor neurons by Ia afferents had reduced amplitude. The number of Ia synapses per motor neurons was unaltered, suggesting that the proprioceptive synapses were weaker in the mutant. Despite the altered stretch reflex amplitude, the adult 5-ht1d-/mice showed normal behavior on an accelerating rotarod. Remarkably, in a beam walking test 5-ht1d-/- mice had fewer foot faults than 5-ht1d+/+ mice suggesting improved coordination abilities. Paper IV Some markers with specific expression in the ventral spinal cord identified in paper I were not expressed in motor neurons. In paper IV, one of these molecular markers was further analyzed. Cholinergic receptor alpha 2 (Chrna2) was expressed in small interneurons in the ventral-most spinal cord that co-expressed the Ca2+-binding protein Calbindin, a previously described Renshaw cell marker [132]. A Chrna2::Cre mouse was created to further study the Chrna2-expressing cells. Patch-clamp recording from Chrna2::Cre;lsl.Tomato-expressing cells showed that these cells responded with a monosynaptic latency to antidromic stimulation of the ventral root. In addition, Chrna2::Cre;lsl.Tomato-expressing axons contacted motor neurons with synapses expressing vesicular inhibitory amino acid transporter (Viaat). These observations suggested that Chrna2 was specific for Renshaw cells. To examine the role of Renshaw cells in development of spinal motor circuits the Chrna2::Cre mouse was crossed with a mouse with a floxed allele for Viaat. The VIAAT protein transports GABA and glycine into synaptic vesicles so tentatively in Chrna2::Cre;Viaatlx/lx mice, Renshaw cells have deficient neurotransmission. In these mutants, whole-cell patch clamp recordings showed that motor neurons developed with increased action potential threshold and decreased action potential amplitude. Motor neurons also had reduced capacity to adapt to increasing current injections with a lower slope of frequency. Other electrophysiological characters such as input resistance, rheobase and AHP half-decay were normal. In addition, motor neurons were also contacted by an increased number of Calbindin+, ChAT+ and VGLUT1+ synapses. However, measurement of the physiology of VGLUT1+ synapses by recording the stretch reflex response showed that despite the increased synaptic number, the stretch reflex amplitude was normal in Chrna2::Cre;Viaatlx/lx mice. 35 Drug-induced fictive locomotion was also normal in the mutant and application of nicotinic antagonist Mecamylamine, which blocks the activation of Renshaw cells by motor neurons, had similar effects in both mutant and control. In addition, analysis of motor behavior of the adult Chrna2::Cre;Viaatlx/lx mice showed no difference including the ability to maintain balance at different speeds (steady rotarod), ability to maintain balance as speed increases (accelerating rotarod), and ability for correct foot placement balance (beam walking). No difference was seen in stride length, stance and swing phase, steeping frequency or other gait parameters either. 36 Discussion Novel markers for subtypes of motor neurons and premotor interneurons Figure 9. Novel molecular markers for subtypes of motor neurons and premotor interneurons presented in this thesis. RC, Renshaw cell. F, fast alpha motor neurons. S?, putative slow alpha motor neuron. , gamma motor neuron. P, partition cell. PN, proprioceptive afferent neuron In this thesis, novel markers are presented for fast alpha motor neurons, gamma motor neurons, Renshaw cells, putative slow motor neurons and medial partition cells (Figure 9). The identification of these genes forms the foundation on which the rest of the work in this thesis stands on. Identification of genes expressed by specific cell populations are valuable for studies 37 of the function of a cell, both as a tool to drive expression of transgenic tools, as a marker of a cell population in histology and by studying the function of the protein. Fast and slow motor neurons In paper I, it is presented that Chodl is expressed in a subtype of large motor neurons. Patch-clamp recordings from CHODL-expressing cells show that they have a shorter AHP half-decay and a larger rheobase current. These are features suggesting that Chodl is expressed in fast motor neurons [18, 19, 133, 134]. Data is also presented that Calcitonin/Calcitonin gene related polypeptide alpha (Calca) is expressed in the same motor neurons as Chodl. The Calca mRNA is spliced into the neuropeptide CGRP in the brain [135]. Observation has been made that CGRP is expressed at NMJs on fast muscle fibers in rats, and is specifically enriched in NMJs innervating fast fatigable muscle fibers [136]. From the data presented in paper I no conclusion on the subtype-specificity of Calca/Chodl-expression in fast motor neurons can be drawn. Thus, Calca and Chodl mark a population of fast motor neurons in the mouse spinal cord. ERR is expressed in a complementary motor neuron population to Chodl and Calca with a small soma area. Expression of ERR mRNA persists in Egr3-/- suggesting that ERR is not expressed by gamma motor neurons. Based on these observation it is suggested in paper I that ERR is expressed by slow motor neurons. However, in light of the recent finding that beta motor neurons persist in the Egr3-/- knockout it cannot be ruled out that ERR is expressed by this motor neuron cell population as well. Expression of ERR is detected in motor neurons already from E11.5 and Chodl is detected in the motor neuron region already from E10.5. Despite extensive studies on motor neuron development over the past two decades, the mechanisms inducing the fast and slow character of motor neurons are largely unknown. The identification of ERR and Chodl expressed in this pattern at this time point of development may provide clues into fast/slow differentiation. ERR is an orphan nuclear receptor that has a role in the differentiation of rod photoreceptors and endolymph-producing epithelia in the inner ear [137, 138]. Notably, in the photoreceptors of ERR knockouts, expression of several genes involved in energy metabolism as well as in ion transport is downregulated, and in the inner ear ion channels and transporters are downregulated. These genes encode two neural characters, membrane properties and metabolic load, where fast and slow motor neurons are different and would be expected to express a different set of genes [139, 140]. The function of the CHODL protein is currently unknown. Chodl mRNA is alternatively spliced into transmembrane or secreted isoforms that all carry a C-type lectin domain [141, 142]. Proteins with C-type lectin domains have been implicated in cell migration and cell-cell interaction [143], and CLEC38 38, a Caenorhabditis elegans with a c-type lectin, modulates unc-40/DCCmediated axon outgrowth and presynaptic patterning [144]. Thus, one potential function of CHODL could be the axon guidance of motor neurons. Gamma motor neurons 5-ht1d is proposed to be specifically expressed by gamma motor neurons based on the following criteria of known gamma motor neuron character [145]: 1. Expression in the smallest motor neurons 2. Expression at all levels of the spinal cord, 3. Loss of expression in Egr3-/- spinal cord. 4. 5-ht1d is expressed by 28% of motor neurons, which is the percentage of motor neurons previously suggested to have a gamma identity [146, 147]. Recently, ERR (also known as ERR3) has been suggested to be specific for gamma motor neurons based on soma size, diminished expression in a mutant mouse which lacks muscle spindles, similar to Egr3-/- mutants and lack of VGLUT1 synapses on the cell body [146]. In addition, through a combination of transgenic mice expressing tau-LacZ from the locus of GDNF receptor GFR 1 (GFR 1-TLZ) and GFP from the Hb9-promoter (Hb9::GFP) gamma motor neurons can be identified as those expressing GFR 1-TLZ while not expressing Hb9::GFP. Again, this was based on the expression in small motor neurons, the diminished expression in Egr3-/- spinal cord and the lack of VGLUT1 input [147]. In contrast to ERR and GFR 1-TLZ/Hb9::GFP, 5-ht1d appears to be specifically expressed by gamma motor neurons throughout postnatal development. ERR is expressed by most motor neurons until the second postnatal week when it becomes specific for gamma motor neurons and GFR 1TLZ is expressed in beta motor neurons and alpha motor neurons as well. 5ht1d is specific for gamma motor neurons at P11 and 5-ht1d::GFP is expressed in small motor neurons from P0 and onwards. 5-ht1d mRNA could not be detected in the spinal cord embryonically or at P0 while a robust expression was seen in the DRGs. Nevertheless, 5-ht1d has not been detected in alpha motor neurons at any stage analyzed, which makes it a more suitable marker for transgenic studies of gamma motor neurons. This is illustrated in paper III where the specific expression of 5-ht1d::GFP allows patch-clamp recordings from gamma motor neurons. Both alpha and gamma motor neurons receive serotonergic input and at least alpha motor neurons express many subtypes of serotonin receptors [148, 149]. Depending on the subtype, activation of serotonin receptors lead to distinct downstream signaling events. Activation of 5-HT1D leads to signaling using the Gi/Go system of G-protein coupled receptors (GPCR) that lead to a reduced excitation through reduced levels of cAMP. However, if this is the effect of 5-HT1D activation on gamma motor neurons remains to be experimentally determined. 39 Renshaw cells Chrna2 is proposed to be specific for Renshaw cells based on the following criteria for a Renshaw cell character [150]: 1. Expression in the ventral grey matter of the spinal cord close to the white matter border, 2. Co-expression with Calbindin, Viaat and large Gephyrin clusters, 3. A mecamylaminesensitive, short-latency monosynaptic response to ventral root stimulation, 4. VIAAT- and Calbindin-expressing synapses originating from Chrna2:Creexpressing cells on motor neurons. Previously, Calbindin and lineage tracing in En1-Cre mice have been the most specific ways to genetically mark Renshaw cell, but these two approaches will also label many non-Renshaw interneurons, likely with integral functions for locomotor control. Therefore, the identification of Chrna2 provides a novel specific way of targeting Renshaw cells for studies of their function. This is illustrated in paper IV in this thesis, where the specific expression of Chrna2 is used to specifically remove expression of the vesicular inhibitory amino acid transporter (Viaat) from Renshaw cells. VIAAT pumps GABA and glycine into synaptic vesicles of inhibitory neurons [151]. Cultured neurons from spinal cord of a full knockout and from hypothalamus in a hypothalamus-specific knockout fail to produce IPSCs suggesting that a knockout of VIAAT is enough to diminish neurotransmission with GABA or glycine [114, 151]. Whether Renshaw cells are deficient in inhibitory neurotransmission has not been assessed by the current study, but extending from the described studies, we consider it likely that Renshaw cell do not signal with inhibitory neurotransmitters in Chrna2::Cre;Viaatlx/lx mice [114, 151]. Partition cells Partition cells are a cell population of cholinergic cells adjacent to the central canal [152]. Pitx2 is proposed to be specifically expressed in partition cells based on the following observations: 1. Large cholinergic neurons close to the central canal that does not co-stain with a retrograde tracer from the ventral root, 2. They form synapses on motor neurons. Another study was published when paper I was under review, came to the same conclusion about Pitx2 neuron identity [22]. Zagoraiou et al. establish that Pitx2 neurons are the source of the C boutons on alpha motor neurons and that this input is required for the task-dependent modulation of muscle activation amplitude. Excitotoxicity and motor neuron subtypes in ALS Reduced Vglut2 expression did not alter the time of disease onset or death in the Sod1G93A mouse model of ALS. However, motor neurons and their con40 nection to the NMJ were partially rescued in the spinal cord and fully rescued in the facial nucleus. These observations are similar to what has been observed previously when extracellular glutamate uptake by astrocytes was increased by overexpressing the plasma membrane transporter of glutamate, EAAT-2 [153]. Thus, reducing glutamate affects motor neuron survival, but does not have a major influence on the general features of disease progression. In the spinal cord of Sod1G93A mice and Sod1G93A;Vglut2+/ mice, expression of fast motor neuron marker Chodl was not detected, while putative slow motor neuron marker ERR was reduced in similar ratios as the general motor neurons markers VAChT and Cht-1. In contrast to the total absence of Chodl expression, few cells in the motor neuron region expressed Calca in both mutants. Since Calca and Chodl are expressed by the same subtype of fast motor neurons, the selective susceptibility of fast motor axon is not directly translated to gene expression in the spinal cord. Both Chodl and Calca mRNA are subject to alternative splicing [135, 142]. Mutations in RNA-editing enzymes TDP-43 and FUS/TLS has been described in cases of familial ALS, suggesting that aberrant mRNA processing may be a common link in the pathogenesis of different mutations in ALS [154]. Notably, aberrant mRNA processing of Chodl mRNA is also seen in motor neurons of a mouse model of another motor neuron degenerative disease, spinal muscular atrophy [155]. Thus, the absence of Chodl mRNA in Sod1G93A mice may be a reflection of hypersensitivity of this mRNA to deficiencies in mRNA processing. Motor neurons in the brain stem appear to be differently affected by disease in Sod1G93A mice [156]. In Sod1G93A;Vglut2+/ mice, the loss of motor neurons in the facial nucleus was completely rescued. The facial motor neurons innervate muscles responsible for facial expressions and do not have muscle spindles. Therefore, they also likely lack the Ia afferents, which provide monosynaptic feedback on muscle stretch directly to the motor neurons [157]. In the rat spinal cord, monosynaptic synapses use VGLUT1 for synaptic transmission and most VGLUT1 input to motor neurons comes from monosynaptic afferents [158]. If facial motor neurons lack this glutamatergic monosynaptic input, it is plausible that reducing expression of VGLUT2 will have a greater impact on reducing glutamate-induced excitotoxicity, which could explain why we see a complete rescue of these motor neurons. 41 The influence of 5-ht1d and VIAAT-mediated Renshaw cell signaling on the development of spinal motor circuits Apart from their role as signaling molecules in neurotransmission in the adult CNS, neurotransmitters are involved in development of neural circuits [159, 160]. The roles neurotransmitter have during development range from developmentally early events such as stem cell proliferation [161] and axon guidance [162, 163] to later roles such as the activity-dependent refinement of neural circuits [95]. In paper III and paper IV we interfere with neurotransmitter signaling in two different ways: in paper III a serotonin receptor is removed from all cells that express it and in paper IV we remove vesicular packaging of GABA and glycine in a small defined population. Both these deletions have implications on motor circuits that suggest a role for them during development. Development of the stretch reflex in 5-ht1d-/- knockout mice In paper III, we demonstrate that full knockouts of 5-ht1d have an almost 50% reduction of the maximal amplitude in the stretch reflex circuit. In 5ht1d-/- mice, Ia afferents form normal annular innervation on muscle spindles in tibialis anterior and have a normal number of VGLUT1+ Ia synapses on the soma and proximal dendrites of motor neurons. Thus, in the absence of 5-ht1d, the stretch reflex circuit is anatomically normal while it is defective in its physiology. Reduced amplitude of the stretch reflex despite normal amounts of Iamotor neuron synapses can have many causes. These include a lower number of motor neurons, hypoexcitable motor neurons, increased short-term depression and reduced synaptic strength in the Ia-motor neuron synapse. The exact number of motor neurons in 5-ht1d mutant mice has not been counted. However, if a 50% reduction in amplitude were caused by a matching 50% reduction in number of motor neurons, it would be clearly noticeable, as the number of motor neurons per LMC in the segments stained is approximately 20-30 in a 60 m section. In addition, in soma area estimations we did not find a depletion of any specific subtype, as the histogram of soma area distributions was the same between 5-ht1d-/- and 5-ht1d+/+ mice. Therefore, it is improbable that a reduced number of motor neurons are the cause of the observed reduction of amplitude. Another possible explanation for the reduced amplitude is that motor neurons develop into a hypoexcitable state in 5-HT1D knockouts. Depletion of serotonin with PCPA for a short period during postnatal development lead to an increased input conductance and an increased rheobase, which are signs of hypoexcitability [164]. The effect of PCPA was only observed in some 42 motor pools as others developed normally. However, in contrast to many other serotonin receptors, 5-HT1D is not expressed on alpha motor neurons. Therefore, if alpha motor neurons were hypoexcitable in 5-ht1d-/- mice, it would be by an indirect mechanism, which makes it less likely. Figure 10. Two hypotheses of reduced stretch reflex amplitude in 5-ht1d-/- mice. (A) In peripheral 5-HT hypothesis, serotonin released by muscle spindles regulates the development of the stretch reflex by signaling via 5-HT1D receptors expressed by Ia proprioceptive neurons. In 5-ht1d-/- mice, this signaling is absent leading to altered development of stretch reflex amplitude. (B) In short-term depression hypothesis, a train of action potentials in Ia afferents (>0.1 Hz) activates a short-term depression of the Ia afferent-motor neuron synapse. This depression mechanism acts via descending 5-HT neuron that presynaptically signals to Ia afferents. In 5-ht1d-/mice, this signaling is absent leading to increased depression of stretch reflex amplitude. The monosynaptic EPSP generated in motor neurons upon dorsal rootstimulation is short-term depressed when stimulated at >0.1 Hz [119]. The exact mechanism behind this depression has not been well described although it involves presynaptic inhibition of Ia afferents and does not involve signaling with GABA [119]. In the stretch reflex experiments in paper III, dorsal root afferents were stimulated at 0.5 Hz, which means this short-term depression mechanism was likely in effect (Figure 10). In the age groups used (P2-P6), stimulus at 0.5 Hz depresses the amplitude by 50% [119]. Serotonin is known to presynaptically inhibit glutamate-release onto motor neurons and depress the stretch reflex [165-168]. This depression is blocked when adding BRL-15572, a selective 5-HT1D antagonist, while BRL-15572 alone has no effect on the amplitude [169]. These results taken together with 43 the observed expression pattern of 5-ht1d in proprioceptive sensory neurons suggest that serotonin could depress the stretch reflex by acting presynaptically on Ia afferents. From these observations, it is possible that the reduced amplitude observed in 5-ht1d-/- was an effect of altered short-term depression in the Ia afferent-motor neurons synapse (Figure 10). A fourth possible explanation is that peripheral serotonin released from the muscle spindle might be involved in synapse development of the Iamotor neuron synapse (Figure 10). Synapse development of the Ia-motor neuron synapse involves signaling both centrally in the CNS and peripherally in the muscle. Muscle spindle-derived signals are necessary for tuning the synaptic strength of the Ia-MN synapse [170, 171]. Conditional deletion of ErbB2 from muscle spindles and full knockout of Egr3 lead to abnormal development of muscle spindles and a reduced stretch reflex amplitude [80, 172]. Muscle spindle-specific deletion of Nt-3 has similar but delayed reduction in stretch reflex amplitude [80]. These observations have led to the suggestion that other muscle spindle-derived factors are involved in tuning the stretch reflex circuit. In paper III, expression of Tph1, Aadc and VMAT2 in muscle spindles at postnatal day 2 is presented. Tph1 and Aadc encode the enzymes that synthesize serotonin from L-Tryptophan. Thus, muscle spindles express the components necessary to produce and release serotonin at the time when Ia-motor neuron synapses are shaped. While it is possible that serotonin contribute to the development of the proprioceptive circuitry, the data presented in this thesis does not distinguish between the proposed hypotheses and they are not mutually exclusive. Future experiments are needed to address these questions. Development of spinal motor circuits in the absence of VIAATmediated Renshaw cell signaling In Chrna2::Cre;Viaatlx/lx mice, the output of both the stretch reflex and locomotor CPG is normal in the isolated spinal cord. In contrast to the normal activity in motor circuits, motor neurons develop with an altered character in Chrna2::Cre;Viaatlx/lx mice. Patch-clamp recordings reveal that the difference between membrane potential and action potential threshold is larger than in Chrna2::Cre;Viaatlx/lx mice compared to controls. This means that a larger depolarization of the membrane potential is required of the motor neuron to generate an action potential. This observation suggests that motor neurons in Chrna2::Cre;Viaatlx/lx mice are less excitable. In addition, the synaptic density of glutamatergic, cholinergic and Calbindin+ synaptic contacts is increased on soma and proximal dendrite of motor neurons, which means that more possible synaptic release sites are in contact with motor neurons Chrna2::Viaatlx/lx mice. These two observations taken together, the hypoexcitability and increased synaptic density, are two factors that could 44 compensate for each other and enables the motor neuron output to be normal in Chrna2::Viaatlx/lx mice (Figure 11). In paper IV, we observed an unchanged stretch reflex amplitude, despite increased number of the VGLUT1+ synapses that are specific for the stretch reflex [146, 173]. Figure 11. Model of development of stretch reflex circuit in absence of VIAATmediated Renshaw cell signaling. (Left) Circuit in Viaatlx/lx mouse. RC, Renshaw cell. MN, motor neuron. (Right) Absence of VIAAT-mediated Renshaw cell signaling lead to increased Ia afferent synapses and reduced excitability. These two parameters compensate for each other and create a normal output of the stretch reflex. Blocking GABAA receptors in the chick embryo by in ovo application of Gabazine transiently reduces spontaneous movements that later recover within 12 hours [98]. Notably, when spontaneous neural activity in the spinal cord is perturbed by addition of gabazine, motor neurons respond to the perturbation with a change in cellular excitability and matching increase in quantal mEPSC [97, 98]. Whether the increase in quantal mEPSC is caused by increased number of synaptic contacts is not established by these studies, but it is a plausible mechanism [174]. This is similar to what we observe in Chrna2::Cre;Viaatlx/lx mice. Thus, it is possible that the hypoexcitability of motor neurons and an increased synaptic input are factors that have been modulated by homeostatic plasticity signaling mechanisms to ensure that motor neuron output activity is normal in the absence of VIAAT-mediated Renshaw cell signaling. 45 Motor behavior in 5-ht1d-/- and Chrna2::Cre;Viaatlx/lx mice Experiments performed in reduced preparations such as fictive locomotion and stretch reflex measurements are useful to determine the physiology of small entities of neural circuits. However, in the intact animal many more circuits are participating to generate motor behavior. In paper III and IV, behavioral assays were applied to assess how the molecular and physiological deficiencies were reflected in the behaving animal. The stretch reflex amplitude in locomotion Egr3-/- knockouts and muscle-spindle specific knockouts of Nt-3 and Erbb2 have been described to have a reduced extracellular or EPSP amplitude of the monosynaptic stretch reflex [74, 80, 172]. These mutants all display an ataxic gait and abnormal posture [74, 80, 172]. In light of this data, it is surprising that 5-ht1d mutants, who also have reduced stretch reflex amplitude, do not display any deficiencies in motor coordination. Even when challenged on a rotarod, these mice behave as control animals, and strikingly, when balancing on a beam walking task, they have fewer hindpaw slips compared to controls. However, while 5-ht1d-/- mice have normal muscle spindle innervation and number of proprioceptive synapses on motor neurons, Egr3-/- knockouts, and muscle-spindle specific knockouts of Nt-3 and Erbb2 have abnormal muscle spindle development and/or reduced number of VGLUT1 synapses on motor neurons [74, 80, 172]. In addition, the reduced amplitude reported for these latter mutants is 80-90%, and it is thus plausible that the proprioceptive feedback to motor neurons is severely impaired causing the ataxic gait. In contrast, in 5-ht1d-/- mice, the amplitude reduction is 50% and above severe phenotypic effect threshold suggesting that muscle spindle feedback remains largely functional. Reduced stretch reflex amplitude suggests deficient afferent feedback from muscle spindles. It is surprising that 5-ht1d-/- mice perform better than controls when crossing a narrow beam even though they have reduced stretch reflex amplitude. However, studies in humans of the H-reflex, an indirect measure of the stretch reflex, have shown that the amplitude of this reflex is modulated during different gaits [175]. Notably, while walking over a narrow beam, the H-reflex amplitude is smaller than when walking on a level treadmill [176]. This may seem counter-intuitive, especially since it is known that gamma motor neurons and Ia afferent are more active during beam walking tasks, which would be expected to cause an increased Hreflex [177]. However, the stretch reflex feedback to motor neurons is known to have a destabilizing effect if overly excited [178, 179]. Therefore, Llewellyn et al. suggested that the Ia afferent-motor neurons connection is 46 presynaptically inhibited during difficult tasks such as beam walking to not overexcite motor neurons, biasing the signaling of Ia afferents to active Ia inhibitory interneurons or spinocerebellar neurons instead [176]. Therefore, it is possible that 5-ht1d-/- mutants are “primed” for skilled walking on a balancing beam by having reduced reflex amplitude. It should be noted however, that given the widespread expression of 5-ht1d in the brain, impact on behavior from such areas needs to be considered when interpreting the improved behavior of 5-ht1d-/- mice. Recurrent inhibition in locomotion Even if changes in excitability and synapse number compensate for the absence of Renshaw-cell signaling to generate a functional output of the stretch reflex, Renshaw cells nevertheless do not express the vesicular transporter of GABA and glycine in Chrna2::Cre;Viaatlx/lx mice. Thus, unless they acquire another signaling mechanism in the absence of Viaat, Renshaw cells are not participating in the activity of motor circuits in Chrna2::Cre;Viaatlx/lx mice. It is unlikely that the role of an entire class of interneurons can be compensated for by alterations in excitability and synaptology of motor neurons. The normal fictive locomotion and motor behaviors are for that reason probable to occur in the absence of VIAAT-mediated Renshaw cell signaling. Thus, the activity of Renshaw cells is not an integral part of locomotor circuits generating locomotion. Moreover, their activity is not needed to generate normal gait, motor coordination and generate maximal grip strength. It is still possible that Renshaw cells have a function in a more specific motor behavior not measured in paper IV. The close physiological proximity of Renshaw cells to motor neurons has led to the idea that they have a central role in movement and locomotion. Various roles of Renshaw cells have been proposed from being important in flexion-extension [180] to regulating force generation [150]. It has however also been suggested that Renshaw cells may have a small role in motor control. This latter suggestion is mainly based on the observation that Renshaw cells make weak connections to motor neurons mainly located to the dendrites [181-184]. In light of this, perhaps it is not surprising that Renshaw cells do not appear to have a major function for motor behavior. 47 Acknowledgements It is with deep sincerity that I wish to thank my colleagues at the department and elsewhere. The five years I have spent at the neuroscience department have been great and I am glad I had such great colleagues to share them with. A special thanks to the following people: My supervisor Klas Kullander. Thank you for creating such a great environment to do research in. That perfect mix of a relaxed and friendly attitude with taking research seriously is an unbeatable combination. I have learned a lot about being a scientist from you. The original gang that was in the lab from the beginning. I will always remember those first years with sentimental nostalgia. Nadine, the lab has not been the same since you left. I think that says it all. Anna, you are the smartest person I know and truly an inspiration. Henrik B, your impressive integrity and fairness will forever have an influence on me. Karin, your friendship has made spending time in the lab so much better. Henrik G, thank you for all the fun and great collaborations over the years. Malin, it was always fun to share an office with you. I miss that. Åsa Mackenzie, you are an inspiration as a scientist. Co-authors in the lab: Kia, for bringing invaluable skills to the lab and being such a great person to work with. Chetan, for having the enthusiasm of a child in a candy store whenever new experiments are suggested. Martin, for all the great times both in the lab and elsewhere. Hanna, for your absurdly efficient pragmatism that always gets things done and for that contagious happiness. Sanja, for all that useful knowledge of statistics that suddenly raised the level on our analysis and for never thinking twice about helping out. Co-authors from other labs that have done all those essential experiments we couldn’t do: Patrick Whelan, Stan Nakanishi, Boris Lamotte d’Incamps, Arild Njå, Warren Tourtellotte, Bayle Shanks and Gregor Eichele. Other people in the lab and at the department: Kasia, for being such a genuinely friendly and kind person. Björn, for your genuine interest in science that always results in great conversations and for you immense help with this thesis. Fatima, Lina, Christiane and all the rest of the Kullander, Mac49 kenzie and Leao labs for five great years. Nicole, Greta and others at the department for being great friends. All students that have helped me over the years: Daniel, Smitha, Henrik, Kali, Tobias, Ingrid, Elodie, Emma and Ida. A special thanks to Pierre who finally managed to get in situs working on muscles and Anders who, apart from contributing vital experiments to paper IV made all schematic illustrations in this thesis. Birgitta for doing all that you do and always taking the time to help. Susanne, Jenny, Sussie and Eva for taking care of the mice. Emma, Ulla and Maria, and previous and current Deans at the department Lars Oreland, Håkan Aldskogius and Ted Ebendal for handling administration. A special thanks to Håkan for being my co-supervisor. A big thank you to Sasha Svensson for making the beautiful cover drawing for this thesis. Thank you to my family and friends for being there. A special thank you to Olof for being a great friend and Elin for making my life so much better. 50 References 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 17. Grillner, S., The motor infrastructure: from ion channels to neuronal networks. Nat Rev Neurosci, 2003. 4(7): p. 573-86. Kandel, E.R., J.H. Schwartz, and T.M. Jessell, Principles of neural science. 4 ed. 2000, East Norwalk: Appleton & Lange. 1414. Thach, W., Motor Systems, in Fundamental Neuroscience, W. Thach, Editor. 1999, Academic Press: London. p. 855-863. Albright, T.D., T.M. Jessell, E.R. Kandel, and M.I. Posner, Neural science: a century of progress and the mysteries that remain. Cell, 2000. 100 Suppl: p. S1-55. Dale, H.H., W. Feldberg, and M. Vogt, Release of acetylcholine at voluntary motor nerve endings. J Physiol, 1936. 86(4): p. 353-80. Katz, B., Quantal mechanism of neural transmitter release. Science, 1971. 173(992): p. 123-6. Kandel, E.R., The molecular biology of memory storage: a dialogue between genes and synapses. Science, 2001. 294(5544): p. 1030-8. McHanwell, S. and T.J. Biscoe, The localization of motoneurons supplying the hindlimb muscles of the mouse. Philos Trans R Soc Lond B Biol Sci, 1981. 293(1069): p. 477-508. Bryan, R.N., D.L. Trevino, and W.D. Willis, Evidence for a common location of alpha and gamma motoneurons. Brain Res, 1972. 38(1): p. 193-6. Westbury, D.R., A comparison of the structures of alpha and gamma-spinal motoneurones of the cat. J Physiol, 1982. 325: p. 79-91. Lagerback, P.A., An ultrastructural study of cat lumbosacral gammamotoneurons after retrograde labelling with horseradish peroxidase. J Comp Neurol, 1985. 240(3): p. 256-64. Lagerback, P.A., S. Cullheim, and B. Ulfhake, Electron microscopic observations on the synaptology of cat sciatic gamma-motoneurons after intracellular staining with horseradish peroxidase. Neurosci Lett, 1986. 70(1): p. 23-7. Hellstrom, J., U. Arvidsson, R. Elde, S. Cullheim, and B. Meister, Differential expression of nerve terminal protein isoforms in VAChT-containing varicosities of the spinal cord ventral horn. J Comp Neurol, 1999. 411(4): p. 578-90. Ornung, G., B. Ragnarson, G. Grant, O.P. Ottersen, J. Storm-Mathisen, and B. Ulfhake, Ia boutons to CCN neurones and motoneurones are enriched with glutamate-like immunoreactivity. Neuroreport, 1995. 6(15): p. 1975-80. Burke, R.E., D.N. Levine, P. Tsairis, and F.E. Zajac, 3rd, Physiological types and histochemical profiles in motor units of the cat gastrocnemius. J Physiol, 1973. 234(3): p. 723-48. Engel, A. and C. Franzini-Armstrong, Myology : basic and clinical. 3rd ed. 2004, New York: McGraw-Hill Medical Pub. Division. 2 v. (1960 ). Eccles, J.C., R.M. Eccles, and A. Lundberg, Durations of afterhyperpolarization of motoneurones supplying fast and slow muscles. Nature, 1957. 179(4565): p. 866-8. 51 18. Gardiner, P.F., Physiological properties of motoneurons innervating different muscle unit types in rat gastrocnemius. J Neurophysiol, 1993. 69(4): p. 1160-70. 19. Zengel, J.E., S.A. Reid, G.W. Sypert, and J.B. Munson, Membrane electrical properties and prediction of motor-unit type of medial gastrocnemius motoneurons in the cat. J Neurophysiol, 1985. 53(5): p. 1323-44. 20. Henneman, E., Relation between size of neurons and their susceptibility to discharge. Science, 1957. 126(3287): p. 1345-7. 21. Mendell, L.M., The size principle: a rule describing the recruitment of motoneurons. J Neurophysiol, 2005. 93(6): p. 3024-6. 22. Zagoraiou, L., T. Akay, J.F. Martin, R.M. Brownstone, T.M. Jessell, and G.B. Miles, A cluster of cholinergic premotor interneurons modulates mouse locomotor activity. Neuron, 2009. 64(5): p. 645-62. 23. Dessaud, E., A.P. McMahon, and J. Briscoe, Pattern formation in the vertebrate neural tube: a sonic hedgehog morphogen-regulated transcriptional network. Development, 2008. 135(15): p. 2489-503. 24. Jessell, T.M., Neuronal specification in the spinal cord: inductive signals and transcriptional codes. Nat Rev Genet, 2000. 1(1): p. 20-9. 25. Shirasaki, R. and S.L. Pfaff, Transcriptional codes and the control of neuronal identity. Annu Rev Neurosci, 2002. 25: p. 251-81. 26. Novitch, B.G., A.I. Chen, and T.M. Jessell, Coordinate regulation of motor neuron subtype identity and pan-neuronal properties by the bHLH repressor Olig2. Neuron, 2001. 31(5): p. 773-89. 27. Vallstedt, A., J. Muhr, A. Pattyn, A. Pierani, M. Mendelsohn, M. Sander, T.M. Jessell, and J. Ericson, Different levels of repressor activity assign redundant and specific roles to Nkx6 genes in motor neuron and interneuron specification. Neuron, 2001. 31(5): p. 743-55. 28. Arber, S., B. Han, M. Mendelsohn, M. Smith, T.M. Jessell, and S. Sockanathan, Requirement for the homeobox gene Hb9 in the consolidation of motor neuron identity. Neuron, 1999. 23(4): p. 659-74. 29. Pfaff, S.L., M. Mendelsohn, C.L. Stewart, T. Edlund, and T.M. Jessell, Requirement for LIM homeobox gene Isl1 in motor neuron generation reveals a motor neuron-dependent step in interneuron differentiation. Cell, 1996. 84(2): p. 309-20. 30. Thaler, J., K. Harrison, K. Sharma, K. Lettieri, J. Kehrl, and S.L. Pfaff, Active suppression of interneuron programs within developing motor neurons revealed by analysis of homeodomain factor HB9. Neuron, 1999. 23(4): p. 675-87. 31. Dasen, J.S., A. De Camilli, B. Wang, P.W. Tucker, and T.M. Jessell, Hox repertoires for motor neuron diversity and connectivity gated by a single accessory factor, FoxP1. Cell, 2008. 134(2): p. 304-16. 32. Dasen, J.S., J.P. Liu, and T.M. Jessell, Motor neuron columnar fate imposed by sequential phases of Hox-c activity. Nature, 2003. 425(6961): p. 926-33. 33. Dasen, J.S., B.C. Tice, S. Brenner-Morton, and T.M. Jessell, A Hox regulatory network establishes motor neuron pool identity and target-muscle connectivity. Cell, 2005. 123(3): p. 477-91. 34. Vult von Steyern, F., V. Martinov, I. Rabben, A. Nja, O. de Lapeyriere, and T. Lomo, The homeodomain transcription factors Islet 1 and HB9 are expressed in adult alpha and gamma motoneurons identified by selective retrograde tracing. Eur J Neurosci, 1999. 11(6): p. 2093-102. 35. Kozeka, K. and M. Ontell, The three-dimensional cytoarchitecture of developing murine muscle spindles. Dev Biol, 1981. 87(1): p. 133-47. 52 36. Holley, J.A., C.C. Wimer, and J.E. Vaughn, Quantitative analyses of neuronal development in the lateral motor column of mouse spinal cord. III. Generation and settling patterns of large and small neurons. J Comp Neurol, 1982. 207(4): p. 333-43. 37. Rafuse, V.F., L.D. Milner, and L.T. Landmesser, Selective innervation of fast and slow muscle regions during early chick neuromuscular development. J Neurosci, 1996. 16(21): p. 6864-77. 38. Rafuse, V.F. and L.T. Landmesser, The pattern of avian intramuscular nerve branching is determined by the innervating motoneuron and its level of polysialic acid. J Neurosci, 2000. 20(3): p. 1056-65. 39. Milner, L.D., V.F. Rafuse, and L.T. Landmesser, Selective fasciculation and divergent pathfinding decisions of embryonic chick motor axons projecting to fast and slow muscle regions. J Neurosci, 1998. 18(9): p. 3297-313. 40. Thompson, W.J., K. Condon, and S.H. Astrow, The origin and selective innervation of early muscle fiber types in the rat. J Neurobiol, 1990. 21(1): p. 212-22. 41. Buller, A.J., J.C. Eccles, and R.M. Eccles, Differentiation of fast and slow muscles in the cat hind limb. J Physiol, 1960. 150: p. 399-416. 42. Lomo, T., R.H. Westgaard, and H.A. Dahl, Contractile properties of muscle: control by pattern of muscle activity in the rat. Proc R Soc Lond B Biol Sci, 1974. 187(1086): p. 99-103. 43. Rosen, D.R., T. Siddique, D. Patterson, D.A. Figlewicz, P. Sapp, A. Hentati, D. Donaldson, J. Goto, J.P. O'Regan, H.X. Deng, and et al., Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature, 1993. 362(6415): p. 59-62. 44. Boillee, S., C. Vande Velde, and D.W. Cleveland, ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron, 2006. 52(1): p. 39-59. 45. Bruijn, L.I., M.W. Becher, M.K. Lee, K.L. Anderson, N.A. Jenkins, N.G. Copeland, S.S. Sisodia, J.D. Rothstein, D.R. Borchelt, D.L. Price, and D.W. Cleveland, ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron, 1997. 18(2): p. 327-38. 46. Rothstein, J.D., M. Van Kammen, A.I. Levey, L.J. Martin, and R.W. Kuncl, Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol, 1995. 38(1): p. 73-84. 47. Ganel, R., T. Ho, N.J. Maragakis, M. Jackson, J.P. Steiner, and J.D. Rothstein, Selective up-regulation of the glial Na+-dependent glutamate transporter GLT1 by a neuroimmunophilin ligand results in neuroprotection. Neurobiol Dis, 2006. 21(3): p. 556-67. 48. Pardo, A.C., V. Wong, L.M. Benson, M. Dykes, K. Tanaka, J.D. Rothstein, and N.J. Maragakis, Loss of the astrocyte glutamate transporter GLT1 modifies disease in SOD1(G93A) mice. Exp Neurol, 2006. 201(1): p. 120-30. 49. Rothstein, J.D., S. Patel, M.R. Regan, C. Haenggeli, Y.H. Huang, D.E. Bergles, L. Jin, M. Dykes Hoberg, S. Vidensky, D.S. Chung, S.V. Toan, L.I. Bruijn, Z.Z. Su, P. Gupta, and P.B. Fisher, Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature, 2005. 433(7021): p. 73-7. 50. Dengler, R., A. Konstanzer, G. Kuther, S. Hesse, W. Wolf, and A. Struppler, Amyotrophic lateral sclerosis: macro-EMG and twitch forces of single motor units. Muscle Nerve, 1990. 13(6): p. 545-50. 53 51. Pun, S., A.F. Santos, S. Saxena, L. Xu, and P. Caroni, Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci, 2006. 9(3): p. 408-19. 52. Cole, J.D., Pride and a Daily Marathon. First MIT press edition ed. 1995, London: Gerald Duckworth & Co. Ltd. 53. Lajoie, Y., N. Teasdale, J.D. Cole, M. Burnett, C. Bard, M. Fleury, R. Forget, J. Paillard, and Y. Lamarre, Gait of a deafferented subject without large myelinated sensory fibers below the neck. Neurology, 1996. 47(1): p. 109-15. 54. Proske, U. and S.C. Gandevia, The kinaesthetic senses. J Physiol, 2009. 587(Pt 17): p. 4139-46. 55. Windhorst, U., Muscle proprioceptive feedback and spinal networks. Brain Res Bull, 2007. 73(4-6): p. 155-202. 56. Hunt, C.C., Mammalian muscle spindle: peripheral mechanisms. Physiol Rev, 1990. 70(3): p. 643-63. 57. Hamill, O.P., A new stretch for muscle spindle research. J Physiol, 2010. 588(Pt 4): p. 551-2. 58. Kokkorogiannis, T., Somatic and intramuscular distribution of muscle spindles and their relation to muscular angiotypes. J Theor Biol, 2004. 229(2): p. 263-80. 59. Emonet-Denand, F., Y. Laporte, P.B. Matthews, and J. Petit, On the subdivision of static and dynamic fusimotor actions on the primary ending of the cat muscle spindle. J Physiol, 1977. 268(3): p. 827-61. 60. Brown, A.G., Organization in the spinal cord : the anatomy and physiology of identified neurones. 1981, Berlin: Springer-Verlag. 238. 61. Jankowska, E. and S.A. Edgley, Functional subdivision of feline spinal interneurons in reflex pathways from group Ib and II muscle afferents; an update. Eur J Neurosci, 2010. 32(6): p. 881-93. 62. Jankowska, E. and M.H. Gladden, A positive feedback circuit involving muscle spindle secondaries and gamma motoneurons in the cat. Prog Brain Res, 1999. 123: p. 149-56. 63. Brownstone, R.M. and T.V. Bui, Spinal interneurons providing input to the final common path during locomotion. Prog Brain Res, 2010. 187: p. 81-95. 64. Jankowska, E., Interneuronal relay in spinal pathways from proprioceptors. Prog Neurobiol, 1992. 38(4): p. 335-78. 65. Eccles, J.C., R.M. Eccles, and F. Magni, Central inhibitory action attributable to presynaptic depolarization produced by muscle afferent volleys. J Physiol, 1961. 159: p. 147-66. 66. Rudomin, P. and R.F. Schmidt, Presynaptic inhibition in the vertebrate spinal cord revisited. Exp Brain Res, 1999. 129(1): p. 1-37. 67. Marmigere, F. and P. Ernfors, Specification and connectivity of neuronal subtypes in the sensory lineage. Nat Rev Neurosci, 2007. 8(2): p. 114-27. 68. Chen, A.I., J.C. de Nooij, and T.M. Jessell, Graded activity of transcription factor Runx3 specifies the laminar termination pattern of sensory axons in the developing spinal cord. Neuron, 2006. 49(3): p. 395-408. 69. Levanon, D., D. Bettoun, C. Harris-Cerruti, E. Woolf, V. Negreanu, R. Eilam, Y. Bernstein, D. Goldenberg, C. Xiao, M. Fliegauf, E. Kremer, F. Otto, O. Brenner, A. Lev-Tov, and Y. Groner, The Runx3 transcription factor regulates development and survival of TrkC dorsal root ganglia neurons. Embo J, 2002. 21(13): p. 3454-63. 70. Wang, G. and S.A. Scott, The "waiting period" of sensory and motor axons in early chick hindlimb: its role in axon pathfinding and neuronal maturation. J Neurosci, 2000. 20(14): p. 5358-66. 54 71. Landmesser, L. and M.G. Honig, Altered sensory projections in the chick hind limb following the early removal of motoneurons. Dev Biol, 1986. 118(2): p. 511-31. 72. Hippenmeyer, S., N.A. Shneider, C. Birchmeier, S.J. Burden, T.M. Jessell, and S. Arber, A role for neuregulin1 signaling in muscle spindle differentiation. Neuron, 2002. 36(6): p. 1035-49. 73. Albert, Y., J. Whitehead, L. Eldredge, J. Carter, X. Gao, and W.G. Tourtellotte, Transcriptional regulation of myotube fate specification and intrafusal muscle fiber morphogenesis. J Cell Biol, 2005. 169(2): p. 257-68. 74. Tourtellotte, W.G. and J. Milbrandt, Sensory ataxia and muscle spindle agenesis in mice lacking the transcription factor Egr3. Nat Genet, 1998. 20(1): p. 87-91. 75. Frank, E. and B. Mendelson, Specification of synaptic connections mediating the simple stretch reflex. J Exp Biol, 1990. 153: p. 71-84. 76. Mendelson, B. and E. Frank, Specific monosynaptic sensory-motor connections form in the absence of patterned neural activity and motoneuronal cell death. J Neurosci, 1991. 11(5): p. 1390-403. 77. Pecho-Vrieseling, E., M. Sigrist, Y. Yoshida, T.M. Jessell, and S. Arber, Specificity of sensory-motor connections encoded by Sema3e-Plxnd1 recognition. Nature, 2009. 459(7248): p. 842-6. 78. Gallego, R., M. Kuno, R. Nunez, and W.D. Snider, Disuse enhances synaptic efficacy in spinal mononeurones. J Physiol, 1979. 291: p. 191-205. 79. Manabe, T., S. Kaneko, and M. Kuno, Disuse-induced enhancement of Ia synaptic transmission in spinal motoneurons of the rat. J Neurosci, 1989. 9(7): p. 2455-61. 80. Shneider, N.A., G.Z. Mentis, J. Schustak, and M.J. O'Donovan, Functionally reduced sensorimotor connections form with normal specificity despite abnormal muscle spindle development: the role of spindle-derived neurotrophin 3. J Neurosci, 2009. 29(15): p. 4719-35. 81. Renshaw, B., Influence of the discharge of motoneurons upon excitation of neighboring motoneurons. J Neurophysiol, 1941. 4: p. 167-183. 82. Renshaw, B., Central effects of centripetal impulses in axons of spinal ventral roots. J Neurophysiol, 1946. 9: p. 191-204. 83. Eccles, J.C., P. Fatt, and K. Koketsu, Cholinergic and inhibitory synapses in a pathway from motor-axon collaterals to motoneurones. J Physiol, 1954. 126(3): p. 524-62. 84. Lamotte d'Incamps, B. and P. Ascher, Four excitatory postsynaptic ionotropic receptors coactivated at the motoneuron-Renshaw cell synapse. J Neurosci, 2008. 28(52): p. 14121-31. 85. Mentis, G.Z., F.J. Alvarez, A. Bonnot, D.S. Richards, D. Gonzalez-Forero, R. Zerda, and M.J. O'Donovan, Noncholinergic excitatory actions of motoneurons in the neonatal mammalian spinal cord. Proc Natl Acad Sci U S A, 2005. 102(20): p. 7344-9. 86. Nishimaru, H., C.E. Restrepo, J. Ryge, Y. Yanagawa, and O. Kiehn, Mammalian motor neurons corelease glutamate and acetylcholine at central synapses. Proc Natl Acad Sci U S A, 2005. 102(14): p. 5245-9. 87. Schneider, S.P. and R.E. Fyffe, Involvement of GABA and glycine in recurrent inhibition of spinal motoneurons. J Neurophysiol, 1992. 68(2): p. 397-406. 88. Windhorst, U., On the role of recurrent inhibitory feedback in motor control. Prog Neurobiol, 1996. 49(6): p. 517-87. 55 89. Sapir, T., E.J. Geiman, Z. Wang, T. Velasquez, S. Mitsui, Y. Yoshihara, E. Frank, F.J. Alvarez, and M. Goulding, Pax6 and engrailed 1 regulate two distinct aspects of renshaw cell development. J Neurosci, 2004. 24(5): p. 1255-64. 90. Saueressig, H., J. Burrill, and M. Goulding, Engrailed-1 and netrin-1 regulate axon pathfinding by association interneurons that project to motor neurons. Development, 1999. 126(19): p. 4201-12. 91. Wenner, P. and M.J. O'Donovan, Identification of an interneuronal population that mediates recurrent inhibition of motoneurons in the developing chick spinal cord. J Neurosci, 1999. 19(17): p. 7557-67. 92. Xu, H., P.J. Whelan, and P. Wenner, Development of an inhibitory interneuronal circuit in the embryonic spinal cord. J Neurophysiol, 2005. 93(5): p. 2922-33. 93. Arai, Y., G.Z. Mentis, J.Y. Wu, and M.J. O'Donovan, Ventrolateral origin of each cycle of rhythmic activity generated by the spinal cord of the chick embryo. PLoS One, 2007. 2(5): p. e417. 94. Wenner, P. and M.J. O'Donovan, Mechanisms that initiate spontaneous network activity in the developing chick spinal cord. J Neurophysiol, 2001. 86(3): p. 1481-98. 95. Blankenship, A.G. and M.B. Feller, Mechanisms underlying spontaneous patterned activity in developing neural circuits. Nat Rev Neurosci, 2010. 11(1): p. 18-29. 96. Gonzalez-Islas, C. and P. Wenner, Spontaneous network activity in the embryonic spinal cord regulates AMPAergic and GABAergic synaptic strength. Neuron, 2006. 49(4): p. 563-75. 97. Wilhelm, J.C., M.M. Rich, and P. Wenner, Compensatory changes in cellular excitability, not synaptic scaling, contribute to homeostatic recovery of embryonic network activity. Proc Natl Acad Sci U S A, 2009. 106(16): p. 6760-5. 98. Wilhelm, J.C. and P. Wenner, GABAA transmission is a critical step in the process of triggering homeostatic increases in quantal amplitude. Proc Natl Acad Sci U S A, 2008. 105(32): p. 11412-7. 99. Hanson, M.G. and L.T. Landmesser, Characterization of the circuits that generate spontaneous episodes of activity in the early embryonic mouse spinal cord. J Neurosci, 2003. 23(2): p. 587-600. 100. Hanson, M.G. and L.T. Landmesser, Normal patterns of spontaneous activity are required for correct motor axon guidance and the expression of specific guidance molecules. Neuron, 2004. 43(5): p. 687-701. 101. Myers, C.P., J.W. Lewcock, M.G. Hanson, S. Gosgnach, J.B. Aimone, F.H. Gage, K.F. Lee, L.T. Landmesser, and S.L. Pfaff, Cholinergic input is required during embryonic development to mediate proper assembly of spinal locomotor circuits. Neuron, 2005. 46(1): p. 37-49. 102. Mentis, G.Z., V.C. Siembab, R. Zerda, M.J. O'Donovan, and F.J. Alvarez, Primary afferent synapses on developing and adult Renshaw cells. J Neurosci, 2006. 26(51): p. 13297-310. 103. Delpy, A., A.E. Allain, P. Meyrand, and P. Branchereau, NKCC1 cotransporter inactivation underlies embryonic development of chloride-mediated inhibition in mouse spinal motoneuron. J Physiol, 2008. 586(4): p. 1059-75. 104. Zhou, Q., J.P. Renard, G. Le Friec, V. Brochard, N. Beaujean, Y. Cherifi, A. Fraichard, and J. Cozzi, Generation of fertile cloned rats by regulating oocyte activation. Science, 2003. 302(5648): p. 1179. 105. John, H.A., M.L. Birnstiel, and K.W. Jones, RNA-DNA hybrids at the cytological level. Nature, 1969. 223(5206): p. 582-7. 56 106. Pardue, M.L. and J.G. Gall, Molecular hybridization of radioactive DNA to the DNA of cytological preparations. Proc Natl Acad Sci U S A, 1969. 64(2): p. 600-4. 107. Liu, W., J. Selever, M.F. Lu, and J.F. Martin, Genetic dissection of Pitx2 in craniofacial development uncovers new functions in branchial arch morphogenesis, late aspects of tooth morphogenesis and cell migration. Development, 2003. 130(25): p. 6375-85. 108. Hippenmeyer, S., E. Vrieseling, M. Sigrist, T. Portmann, C. Laengle, D.R. Ladle, and S. Arber, A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLoS Biol, 2005. 3(5): p. e159. 109. Wallen-Mackenzie, A., H. Gezelius, M. Thoby-Brisson, A. Nygard, A. Enjin, F. Fujiyama, G. Fortin, and K. Kullander, Vesicular glutamate transporter 2 is required for central respiratory rhythm generation but not for locomotor central pattern generation. J Neurosci, 2006. 26(47): p. 12294-307. 110. Gurney, M.E., H. Pu, A.Y. Chiu, M.C. Dal Canto, C.Y. Polchow, D.D. Alexander, J. Caliendo, A. Hentati, Y.W. Kwon, H.X. Deng, and et al., Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science, 1994. 264(5166): p. 1772-5. 111. Lallemand, Y., V. Luria, R. Haffner-Krausz, and P. Lonai, Maternally expressed PGK-Cre transgene as a tool for early and uniform activation of the Cre site-specific recombinase. Transgenic Res, 1998. 7(2): p. 105-12. 112. Lexicon Genetics Inc., NIH initiative supporting placement of Lexicon Genetics, Inc. mice into public repositories. MGI Direct Data Submission, 2005. 113. Gong, S., C. Zheng, M.L. Doughty, K. Losos, N. Didkovsky, U.B. Schambra, N.J. Nowak, A. Joyner, G. Leblanc, M.E. Hatten, and N. Heintz, A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature, 2003. 425(6961): p. 917-25. 114. Tong, Q., C.P. Ye, J.E. Jones, J.K. Elmquist, and B.B. Lowell, Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat Neurosci, 2008. 11(9): p. 998-1000. 115. Madisen, L., T.A. Zwingman, S.M. Sunkin, S.W. Oh, H.A. Zariwala, H. Gu, L.L. Ng, R.D. Palmiter, M.J. Hawrylycz, A.R. Jones, E.S. Lein, and H. Zeng, A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci, 2010. 13(1): p. 133-40. 116. Saper, C.B., An open letter to our readers on the use of antibodies. J Comp Neurol, 2005. 493(4): p. 477-8. 117. Wiedenmann, B. and W.W. Franke, Identification and localization of synaptophysin, an integral membrane glycoprotein of Mr 38,000 characteristic of presynaptic vesicles. Cell, 1985. 41(3): p. 1017-28. 118. Jiang, Z., K.P. Carlin, and R.M. Brownstone, An in vitro functionally mature mouse spinal cord preparation for the study of spinal motor networks. Brain Res, 1999. 816(2): p. 493-9. 119. Li, Y. and R.E. Burke, Developmental changes in short-term synaptic depression in the neonatal mouse spinal cord. J Neurophysiol, 2002. 88(6): p. 3218-31. 120. Kiehn, O. and O. Kjaerulff, Spatiotemporal characteristics of 5-HT and dopamine-induced rhythmic hindlimb activity in the in vitro neonatal rat. J Neurophysiol, 1996. 75(4): p. 1472-82. 121. Bonnot, A., P.J. Whelan, G.Z. Mentis, and M.J. O'Donovan, Locomotor-like activity generated by the neonatal mouse spinal cord. Brain Res Brain Res Rev, 2002. 40(1-3): p. 141-51. 57 122. Gallarda, B.W., T.O. Sharpee, S.L. Pfaff, and W.A. Alaynick, Defining rhythmic locomotor burst patterns using a continuous wavelet transform. Ann N Y Acad Sci, 2010. 1198: p. 133-9. 123. Mor, Y. and A. Lev-Tov, Analysis of rhythmic patterns produced by spinal neural networks. J Neurophysiol, 2007. 98(5): p. 2807-17. 124. Zhang, Y., S. Narayan, E. Geiman, G.M. Lanuza, T. Velasquez, B. Shanks, T. Akay, J. Dyck, K. Pearson, S. Gosgnach, C.M. Fan, and M. Goulding, V3 spinal neurons establish a robust and balanced locomotor rhythm during walking. Neuron, 2008. 60(1): p. 84-96. 125. Dunham, N.W. and T.S. Miya, A note on a simple apparatus for detecting neurological deficit in rats and mice. J Am Pharm Assoc Am Pharm Assoc (Baltim), 1957. 46(3): p. 208-9. 126. Gosgnach, S., G.M. Lanuza, S.J. Butt, H. Saueressig, Y. Zhang, T. Velasquez, D. Riethmacher, E.M. Callaway, O. Kiehn, and M. Goulding, V1 spinal neurons regulate the speed of vertebrate locomotor outputs. Nature, 2006. 440(7081): p. 215-9. 127. Klein, R., I. Silos-Santiago, R.J. Smeyne, S.A. Lira, R. Brambilla, S. Bryant, L. Zhang, W.D. Snider, and M. Barbacid, Disruption of the neurotrophin-3 receptor gene trkC eliminates la muscle afferents and results in abnormal movements. Nature, 1994. 368(6468): p. 249-51. 128. Brooks, S.P. and S.B. Dunnett, Tests to assess motor phenotype in mice: a user's guide. Nat Rev Neurosci, 2009. 10(7): p. 519-29. 129. Wallace, J.E., E.E. Krauter, and B.A. Campbell, Motor and reflexive behavior in the aging rat. J Gerontol, 1980. 35(3): p. 364-70. 130. Helgren, M.E., K.D. Cliffer, K. Torrento, C. Cavnor, R. Curtis, P.S. DiStefano, S.J. Wiegand, and R.M. Lindsay, Neurotrophin-3 administration attenuates deficits of pyridoxine-induced large-fiber sensory neuropathy. J Neurosci, 1997. 17(1): p. 372-82. 131. Wooley, C.M., R.B. Sher, A. Kale, W.N. Frankel, G.A. Cox, and K.L. Seburn, Gait analysis detects early changes in transgenic SOD1(G93A) mice. Muscle Nerve, 2005. 32(1): p. 43-50. 132. Carr, P.A., F.J. Alvarez, E.A. Leman, and R.E. Fyffe, Calbindin D28k expression in immunohistochemically identified Renshaw cells. Neuroreport, 1998. 9(11): p. 2657-61. 133. Burke, R.E., P.L. Strick, K. Kanda, C.C. Kim, and B. Walmsley, Anatomy of medial gastrocnemius and soleus motor nuclei in cat spinal cord. J Neurophysiol, 1977. 40(3): p. 667-80. 134. Ulfhake, B. and J.O. Kellerth, Does alpha-motoneurone size correlate with motor unit type in cat triceps surae? Brain Res, 1982. 251(2): p. 201-9. 135. Rosenfeld, M.G., J.J. Mermod, S.G. Amara, L.W. Swanson, P.E. Sawchenko, J. Rivier, W.W. Vale, and R.M. Evans, Production of a novel neuropeptide encoded by the calcitonin gene via tissue-specific RNA processing. Nature, 1983. 304(5922): p. 129-35. 136. Forsgren, S., A. Bergh, E. Carlsson, and L.E. Thornell, Calcitonin gene-related peptide expression at endplates of different fibre types in muscles in rat hind limbs. Cell Tissue Res, 1993. 274(3): p. 439-46. 137. Chen, J. and J. Nathans, Estrogen-related receptor beta/NR3B2 controls epithelial cell fate and endolymph production by the stria vascularis. Dev Cell, 2007. 13(3): p. 325-37. 138. Onishi, A., G.H. Peng, E.M. Poth, D.A. Lee, J. Chen, U. Alexis, J. de Melo, S. Chen, and S. Blackshaw, The orphan nuclear hormone receptor ERRbeta con- 58 139. 140. 141. 142. 143. 144. 145. 146. 147. 148. 149. 150. 151. 152. 153. 154. trols rod photoreceptor survival. Proc Natl Acad Sci U S A, 2010. 107(25): p. 11579-84. Kernell, D., R. Bakels, and J.C. Copray, Discharge properties of motoneurones: how are they matched to the properties and use of their muscle units? J Physiol Paris, 1999. 93(1-2): p. 87-96. Sickles, D.W. and T.G. Oblak, Metabolic variation among alpha-motoneurons innervating different muscle-fiber types. I. Oxidative enzyme activity. J Neurophysiol, 1984. 51(3): p. 529-37. Weng, L., R. Hubner, A. Claessens, P. Smits, J. Wauters, P. Tylzanowski, E. Van Marck, and J. Merregaert, Isolation and characterization of chondrolectin (Chodl), a novel C-type lectin predominantly expressed in muscle cells. Gene, 2003. 308: p. 21-9. Weng, L., D.R. Van Bockstaele, J. Wauters, E. Van Marck, J. Plum, Z.N. Berneman, and J. Merregaert, A novel alternative spliced chondrolectin isoform lacking the transmembrane domain is expressed during T cell maturation. J Biol Chem, 2003. 278(21): p. 19164-70. Figdor, C.G., Y. van Kooyk, and G.J. Adema, C-type lectin receptors on dendritic cells and Langerhans cells. Nat Rev Immunol, 2002. 2(2): p. 77-84. Kulkarni, G., H. Li, and W.G. Wadsworth, CLEC-38, a transmembrane protein with C-type lectin-like domains, negatively regulates UNC-40-mediated axon outgrowth and promotes presynaptic development in Caenorhabditis elegans. J Neurosci, 2008. 28(17): p. 4541-50. Kanning, K.C., A. Kaplan, and C.E. Henderson, Motor neuron diversity in development and disease. Annu Rev Neurosci, 2010. 33: p. 409-40. Friese, A., J.A. Kaltschmidt, D.R. Ladle, M. Sigrist, T.M. Jessell, and S. Arber, Gamma and alpha motor neurons distinguished by expression of transcription factor Err3. Proc Natl Acad Sci U S A, 2009. 106(32): p. 13588-93. Shneider, N.A., M.N. Brown, C.A. Smith, J. Pickel, and F.J. Alvarez, Gamma motor neurons express distinct genetic markers at birth and require muscle spindle-derived GDNF for postnatal survival. Neural Dev, 2009. 4: p. 42. Gladden, M.H., D.J. Maxwell, A. Sahal, and E. Jankowska, Coupling between serotoninergic and noradrenergic neurones and gamma-motoneurones in the cat. J Physiol, 2000. 527 Pt 2: p. 213-23. Heckman, C.J., C. Mottram, K. Quinlan, R. Theiss, and J. Schuster, Motoneuron excitability: the importance of neuromodulatory inputs. Clin Neurophysiol, 2009. 120(12): p. 2040-54. Alvarez, F.J. and R.E. Fyffe, The continuing case for the Renshaw cell. J Physiol, 2007. 584(Pt 1): p. 31-45. Wojcik, S.M., S. Katsurabayashi, I. Guillemin, E. Friauf, C. Rosenmund, N. Brose, and J.S. Rhee, A shared vesicular carrier allows synaptic corelease of GABA and glycine. Neuron, 2006. 50(4): p. 575-87. Barber, R.P., P.E. Phelps, C.R. Houser, G.D. Crawford, P.M. Salvaterra, and J.E. Vaughn, The morphology and distribution of neurons containing choline acetyltransferase in the adult rat spinal cord: an immunocytochemical study. J Comp Neurol, 1984. 229(3): p. 329-46. Guo, H., L. Lai, M.E. Butchbach, M.P. Stockinger, X. Shan, G.A. Bishop, and C.L. Lin, Increased expression of the glial glutamate transporter EAAT2 modulates excitotoxicity and delays the onset but not the outcome of ALS in mice. Hum Mol Genet, 2003. 12(19): p. 2519-32. Lagier-Tourenne, C. and D.W. Cleveland, Rethinking ALS: the FUS about TDP-43. Cell, 2009. 136(6): p. 1001-4. 59 155. Zhang, Z., F. Lotti, K. Dittmar, I. Younis, L. Wan, M. Kasim, and G. Dreyfuss, SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell, 2008. 133(4): p. 585-600. 156. Haenggeli, C. and A.C. Kato, Differential vulnerability of cranial motoneurons in mouse models with motor neuron degeneration. Neurosci Lett, 2002. 335(1): p. 39-43. 157. Whitehead, J., C. Keller-Peck, J. Kucera, and W.G. Tourtellotte, Glial cell-line derived neurotrophic factor-dependent fusimotor neuron survival during development. Mech Dev, 2005. 122(1): p. 27-41. 158. Oliveira, A.L., F. Hydling, E. Olsson, T. Shi, R.H. Edwards, F. Fujiyama, T. Kaneko, T. Hokfelt, S. Cullheim, and B. Meister, Cellular localization of three vesicular glutamate transporter mRNAs and proteins in rat spinal cord and dorsal root ganglia. Synapse, 2003. 50(2): p. 117-29. 159. Gaspar, P., O. Cases, and L. Maroteaux, The developmental role of serotonin: news from mouse molecular genetics. Nat Rev Neurosci, 2003. 4(12): p. 1002-12. 160. Owens, D.F. and A.R. Kriegstein, Is there more to GABA than synaptic inhibition? Nat Rev Neurosci, 2002. 3(9): p. 715-27. 161. Andang, M., J. Hjerling-Leffler, A. Moliner, T.K. Lundgren, G. CasteloBranco, E. Nanou, E. Pozas, V. Bryja, S. Halliez, H. Nishimaru, J. Wilbertz, E. Arenas, M. Koltzenburg, P. Charnay, A. El Manira, C.F. Ibanez, and P. Ernfors, Histone H2AX-dependent GABA(A) receptor regulation of stem cell proliferation. Nature, 2008. 451(7177): p. 460-4. 162. Bonnin, A., M. Torii, L. Wang, P. Rakic, and P. Levitt, Serotonin modulates the response of embryonic thalamocortical axons to netrin-1. Nat Neurosci, 2007. 10(5): p. 588-97. 163. Lipton, S.A. and S.B. Kater, Neurotransmitter regulation of neuronal outgrowth, plasticity and survival. Trends Neurosci, 1989. 12(7): p. 265-70. 164. Pflieger, J.F., F. Clarac, and L. Vinay, Postural modifications and neuronal excitability changes induced by a short-term serotonin depletion during neonatal development in the rat. J Neurosci, 2002. 22(12): p. 5108-17. 165. Schmidt, B.J. and L.M. Jordan, The role of serotonin in reflex modulation and locomotor rhythm production in the mammalian spinal cord. Brain Res Bull, 2000. 53(5): p. 689-710. 166. Schwartz, E.J., T. Gerachshenko, and S. Alford, 5-HT prolongs ventral root bursting via presynaptic inhibition of synaptic activity during fictive locomotion in lamprey. J Neurophysiol, 2005. 93(2): p. 980-8. 167. Singer, J.H., M.C. Bellingham, and A.J. Berger, Presynaptic inhibition of glutamatergic synaptic transmission to rat motoneurons by serotonin. J Neurophysiol, 1996. 76(2): p. 799-807. 168. Wu, S.Y., M.Y. Wang, and N.J. Dun, Serotonin via presynaptic 5-HT1 receptors attenuates synaptic transmission to immature rat motoneurons in vitro. Brain Res, 1991. 554(1-2): p. 111-21. 169. Honda, M., K. Imaida, M. Tanabe, and H. Ono, Endogenously released 5hydroxytryptamine depresses the spinal monosynaptic reflex via 5-HT1D receptors. Eur J Pharmacol, 2004. 503(1-3): p. 55-61. 170. Frank, E. and P. Wenner, Environmental specification of neuronal connectivity. Neuron, 1993. 10(5): p. 779-85. 171. Mentis, G.Z., F.J. Alvarez, N.A. Shneider, V.C. Siembab, and M.J. O'Donovan, Mechanisms regulating the specificity and strength of muscle afferent inputs in the spinal cord. Ann N Y Acad Sci, 2010. 1198: p. 220-30. 60 172. Chen, H.H., W.G. Tourtellotte, and E. Frank, Muscle spindle-derived neurotrophin 3 regulates synaptic connectivity between muscle sensory and motor neurons. J Neurosci, 2002. 22(9): p. 3512-9. 173. Betley, J.N., C.V. Wright, Y. Kawaguchi, F. Erdelyi, G. Szabo, T.M. Jessell, and J.A. Kaltschmidt, Stringent specificity in the construction of a GABAergic presynaptic inhibitory circuit. Cell, 2009. 139(1): p. 161-74. 174. Pratt, K.G., A.J. Watt, L.C. Griffith, S.B. Nelson, and G.G. Turrigiano, Activity-dependent remodeling of presynaptic inputs by postsynaptic expression of activated CaMKII. Neuron, 2003. 39(2): p. 269-81. 175. Edamura, M., J.F. Yang, and R.B. Stein, Factors that determine the magnitude and time course of human H-reflexes in locomotion. J Neurosci, 1991. 11(2): p. 420-7. 176. Llewellyn, M., J.F. Yang, and A. Prochazka, Human H-reflexes are smaller in difficult beam walking than in normal treadmill walking. Exp Brain Res, 1990. 83(1): p. 22-8. 177. Hulliger, M., N. Durmuller, A. Prochazka, and P. Trend, Flexible fusimotor control of muscle spindle feedback during a variety of natural movements. Prog Brain Res, 1989. 80: p. 87-101; discussion 57-60. 178. Cussons, P.D., P.B. Matthews, and R.B. Muir, Enhancement by agonist or antagonist muscle vibration of tremor at the elastically loaded human elbow. J Physiol, 1980. 302: p. 443-61. 179. Rack, P.M., H.F. Ross, and A.F. Thilmann, The ankle stretch reflexes in normal and spastic subjects. The response to sinusoidal movement. Brain, 1984. 107 ( Pt 2): p. 637-54. 180. Miller, S. and P.D. Scott, The spinal locomotor generator. Exp Brain Res, 1977. 30(2-3): p. 387-403. 181. Fyffe, R.E., Spatial distribution of recurrent inhibitory synapses on spinal motoneurons in the cat. J Neurophysiol, 1991. 65(5): p. 1134-49. 182. Hamm, T.M., S. Sasaki, D.G. Stuart, U. Windhorst, and C.S. Yuan, Distribution of single-axon recurrent inhibitory post-synaptic potentials in a single spinal motor nucleus in the cat. J Physiol, 1987. 388: p. 653-64. 183. Lindsay, A.D. and M.D. Binder, Distribution of effective synaptic currents underlying recurrent inhibition in cat triceps surae motoneurons. J Neurophysiol, 1991. 65(2): p. 168-77. 184. Maltenfort, M.G., M.L. McCurdy, C.A. Phillips, V.V. Turkin, and T.M. Hamm, Location and magnitude of conductance changes produced by Renshaw recurrent inhibition in spinal motoneurons. J Neurophysiol, 2004. 92(3): p. 1417-32. 61