
Sequence Similarity Searching: Understanding and Using Web
... • A cycling/iterative method – Gives increased sensitivity for detecting distantly related proteins – Can give insight into functional relationships – Very refined statistical methods ...
... • A cycling/iterative method – Gives increased sensitivity for detecting distantly related proteins – Can give insight into functional relationships – Very refined statistical methods ...

Structure and function of haemoglobin: II. Some
... Residues are defined here as invariant when they occur at structurally identical sites in all the normal myoglobins and haemoglobins so far investigated. Abnormal haemoglobins have been excluded, because some of their abnormalities interfere with the oxygen-combining function, so that the protein ca ...
... Residues are defined here as invariant when they occur at structurally identical sites in all the normal myoglobins and haemoglobins so far investigated. Abnormal haemoglobins have been excluded, because some of their abnormalities interfere with the oxygen-combining function, so that the protein ca ...

Diversity of Amyloid Motifs in NLR Signaling in Fungi
... In fungi, when different isolates of the same species fuse, most often a cell death reaction ensues. This phenomenon known as heterokaryon incompatibility is controlled by specific loci termed heterokaryon incompatibility genes (het genes) [12]. In the species Podospora anserina, the het-s (small s) ...
... In fungi, when different isolates of the same species fuse, most often a cell death reaction ensues. This phenomenon known as heterokaryon incompatibility is controlled by specific loci termed heterokaryon incompatibility genes (het genes) [12]. In the species Podospora anserina, the het-s (small s) ...

Supplementary Table 1
... protein necessary for transport from ER to Golgi; required for assembly of the ER-to-Golgi SNARE complex ...
... protein necessary for transport from ER to Golgi; required for assembly of the ER-to-Golgi SNARE complex ...

EP 1790660 B1
... [0020] The method will typically involve the steps of: obtaining nucleic acid encoding a protein of the invention; manipulating said nucleic acid to remove at least one domain from within the protein. The resulting nucleic acid may be inserted into an expression vector, or may already be part of an ...
... [0020] The method will typically involve the steps of: obtaining nucleic acid encoding a protein of the invention; manipulating said nucleic acid to remove at least one domain from within the protein. The resulting nucleic acid may be inserted into an expression vector, or may already be part of an ...

Engineering subunit association of multisubunit proteins
... analysis of complexes, assumes, as a first-order approximation, rigid body association. The same binding free energy evaluation model could also be used to address the third problem for further design of dimeric streptavidins, in which the biotinbinding affinity, reduced due to the subunit separatio ...
... analysis of complexes, assumes, as a first-order approximation, rigid body association. The same binding free energy evaluation model could also be used to address the third problem for further design of dimeric streptavidins, in which the biotinbinding affinity, reduced due to the subunit separatio ...

Comparison of conserved structural and regulatory domains within
... ITS species was found in each strain. This is surprising, ...
... ITS species was found in each strain. This is surprising, ...

Nucleotide sequence and structural organization of
... majority of plasmids have n o known function. The study of small plasmids from a variety of bacteria has recently become a focus of research interest and in several cases this work has led to the development of families of vectors designed for specific purposes. The genetic organization and mode of ...
... majority of plasmids have n o known function. The study of small plasmids from a variety of bacteria has recently become a focus of research interest and in several cases this work has led to the development of families of vectors designed for specific purposes. The genetic organization and mode of ...

Supporting document 1 Safety assessment
... either a coding or regulatory region of the expression cassette and therefore do not have any impact on the expression of the inserted DNA. The transformation event also resulted in a partial duplication of the csr1-2 coding sequence directly before the 3’ integration point, generating a 501 bp open ...
... either a coding or regulatory region of the expression cassette and therefore do not have any impact on the expression of the inserted DNA. The transformation event also resulted in a partial duplication of the csr1-2 coding sequence directly before the 3’ integration point, generating a 501 bp open ...
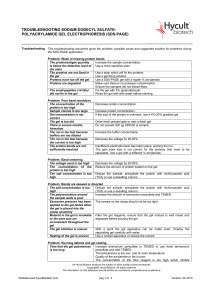
Troubleshooting SDS-PAGE-0410
... There are no net negative charges on proteins, the protein will not move down the gel, ensure SDS has been added to the sample. The solution is acidic, add NaOH until the solution turns blue. There is too little bromophenol blue in the sample buffer. The buffers are too concentrated, dilute the buff ...
... There are no net negative charges on proteins, the protein will not move down the gel, ensure SDS has been added to the sample. The solution is acidic, add NaOH until the solution turns blue. There is too little bromophenol blue in the sample buffer. The buffers are too concentrated, dilute the buff ...


Distinct profiling of antimicrobial peptide families
... potential substitute for conventional antibiotics. Designing new AMPs using current in-silico approaches is, however, challenging due to the absence of suitable models, large number of design parameters, testing cycles, production time and cost. To date, AMPs have merely been categorized into famili ...
... potential substitute for conventional antibiotics. Designing new AMPs using current in-silico approaches is, however, challenging due to the absence of suitable models, large number of design parameters, testing cycles, production time and cost. To date, AMPs have merely been categorized into famili ...

BIOGRAPHICAL SKETCH Joanne I. Yeh joanneyeh Associate
... Glycerol-3-Phosphate Dehydrogenase: a new molecular target in Pseudomonas aeruginosa Major goals of this study are to identify and characterize a new molecular target in P. aeruginosa and to develop inhibitors based on these structural results. No overlap with the proposal being submitted. Role: PI ...
... Glycerol-3-Phosphate Dehydrogenase: a new molecular target in Pseudomonas aeruginosa Major goals of this study are to identify and characterize a new molecular target in P. aeruginosa and to develop inhibitors based on these structural results. No overlap with the proposal being submitted. Role: PI ...

instructions on the annotation of pdf files
... beyond that which is provided by domain length. The respective Fisher’s exact test p values of 0.32 and 0.798 for the case above indicate that the bias in ACO values is not due to differences in domain lengths (Table S1). In the case of relative contact order (RCO) calculations (see Experimental Pro ...
... beyond that which is provided by domain length. The respective Fisher’s exact test p values of 0.32 and 0.798 for the case above indicate that the bias in ACO values is not due to differences in domain lengths (Table S1). In the case of relative contact order (RCO) calculations (see Experimental Pro ...

Journal of Microbiology and Biotechnology
... crystal structure of the IPDC from E. cloacae has recently been determined, and the native enzyme was estimated to be around 240 kDa, suggesting a homotetramer structure [27]. The enzyme is dependent on Mg2+ and thiamine diphosphate (ThDP) as cofactors, and has higher affinity for the substrate IPA ...
... crystal structure of the IPDC from E. cloacae has recently been determined, and the native enzyme was estimated to be around 240 kDa, suggesting a homotetramer structure [27]. The enzyme is dependent on Mg2+ and thiamine diphosphate (ThDP) as cofactors, and has higher affinity for the substrate IPA ...

World Index of BioMolecular Visualization Resources
... Cross-indexing terms: antibody; antibodies; multiple sequence alignment; sequence variability; homology modelling; immunoglobulin; domains; color-coded multiple sequence alignment. This site contains extensive tutorials and lectures on antibody structure coupled with analyses of sequence variability ...
... Cross-indexing terms: antibody; antibodies; multiple sequence alignment; sequence variability; homology modelling; immunoglobulin; domains; color-coded multiple sequence alignment. This site contains extensive tutorials and lectures on antibody structure coupled with analyses of sequence variability ...

Are You suprised ?
... (mutation) of a gene that codes for a protein may result in a change in the amino-acid sequence of the protein. Biochemical evidence of evolution compares favorably with structural evidence of evolution. Even organisms that appear to have few physical similarities may have similar sequences of amino ...
... (mutation) of a gene that codes for a protein may result in a change in the amino-acid sequence of the protein. Biochemical evidence of evolution compares favorably with structural evidence of evolution. Even organisms that appear to have few physical similarities may have similar sequences of amino ...

Protein Purification by Inverse Transition Cycling
... can be easily imparted by genetic fusion to a protein of interest. This is useful because, when the transition is triggered, the fusion protein aggregates and can be collected by centrifugation or filtration. The transition is reversible, and therefore the pelleted ELP fusion protein can be re-solub ...
... can be easily imparted by genetic fusion to a protein of interest. This is useful because, when the transition is triggered, the fusion protein aggregates and can be collected by centrifugation or filtration. The transition is reversible, and therefore the pelleted ELP fusion protein can be re-solub ...

Point Mutations Define a Sequence Flanking the
... that the variability of the assay is 620%. With that in mind, it seems safe to draw conclusions about plasmids that differ 2-fold or more in their production of proinsulin; but when the increment is less than 2-fold, as with 841, the result cannot be considered more than suggestive. The conclusion f ...
... that the variability of the assay is 620%. With that in mind, it seems safe to draw conclusions about plasmids that differ 2-fold or more in their production of proinsulin; but when the increment is less than 2-fold, as with 841, the result cannot be considered more than suggestive. The conclusion f ...

Protein-protein interactions: mechanisms and
... the electrostatic interaction can define the lifetime of complexes (Archakov and Ivanov, 1999). (d) Hydrogen bonding. The average number of hydrogen bonds is proportional to the area of subunit interfaces: one bond for each 100–200 Å2 (Jones and Thornton, 1996) or about 10 bonds per interface (Lo C ...
... the electrostatic interaction can define the lifetime of complexes (Archakov and Ivanov, 1999). (d) Hydrogen bonding. The average number of hydrogen bonds is proportional to the area of subunit interfaces: one bond for each 100–200 Å2 (Jones and Thornton, 1996) or about 10 bonds per interface (Lo C ...

Trade-offs between tRNA abundance and mRNA secondary
... Fast-translated codons are enriched in regions with high secondary structure propensity To assess whether potential trade-offs are made between tRNA abundance and mRNA secondary structure, we first focused on S. cerevisiae for which experimental in vivo mRNA secondary structure measurements are avai ...
... Fast-translated codons are enriched in regions with high secondary structure propensity To assess whether potential trade-offs are made between tRNA abundance and mRNA secondary structure, we first focused on S. cerevisiae for which experimental in vivo mRNA secondary structure measurements are avai ...

Critically discuss the methods that are in routine use for the
... binding is usually measured. However, dye binding methods are less specific than nephelometry or turbidimetry and are inaccurate if the overall serum protein pattern is abnormal, such as in multiple myeloma. The BCG and BCP methods both have advantages and disadvantages, with neither being clearly p ...
... binding is usually measured. However, dye binding methods are less specific than nephelometry or turbidimetry and are inaccurate if the overall serum protein pattern is abnormal, such as in multiple myeloma. The BCG and BCP methods both have advantages and disadvantages, with neither being clearly p ...

Are You suprised ?
... (mutation) of a gene that codes for a protein may result in a change in the amino-acid sequence of the protein. Biochemical evidence of evolution compares favorably with structural evidence of evolution. Even organisms that appear to have few physical similarities may have similar sequences of amino ...
... (mutation) of a gene that codes for a protein may result in a change in the amino-acid sequence of the protein. Biochemical evidence of evolution compares favorably with structural evidence of evolution. Even organisms that appear to have few physical similarities may have similar sequences of amino ...

meme
... present in all of the sequences. * anr — Any Number of Repetitions MEME assumes each sequence may contain any number of non-overlapping occurrences of each motif. This option is useful when you suspect that motifs repeat multiple times within a single sequence. In that case, the motifs found will be ...
... present in all of the sequences. * anr — Any Number of Repetitions MEME assumes each sequence may contain any number of non-overlapping occurrences of each motif. This option is useful when you suspect that motifs repeat multiple times within a single sequence. In that case, the motifs found will be ...

APMLE Practice Test II
... Directions for questions 1-56: These questions are followed by four suggested answers. Select the one answer that is best in each case. NOTE: Throughout this test, the term “medial oblique foot” refers to a nonweightbearing medial oblique position in which the film is flat on the orthoposer, the med ...
... Directions for questions 1-56: These questions are followed by four suggested answers. Select the one answer that is best in each case. NOTE: Throughout this test, the term “medial oblique foot” refers to a nonweightbearing medial oblique position in which the film is flat on the orthoposer, the med ...
Homology modeling

Homology modeling, also known as comparative modeling of protein, refers to constructing an atomic-resolution model of the ""target"" protein from its amino acid sequence and an experimental three-dimensional structure of a related homologous protein (the ""template""). Homology modeling relies on the identification of one or more known protein structures likely to resemble the structure of the query sequence, and on the production of an alignment that maps residues in the query sequence to residues in the template sequence. It has been shown that protein structures are more conserved than protein sequences amongst homologues, but sequences falling below a 20% sequence identity can have very different structure.Evolutionarily related proteins have similar sequences and naturally occurring homologous proteins have similar protein structure.It has been shown that three-dimensional protein structure is evolutionarily more conserved than would be expected on the basis of sequence conservation alone.The sequence alignment and template structure are then used to produce a structural model of the target. Because protein structures are more conserved than DNA sequences, detectable levels of sequence similarity usually imply significant structural similarity.The quality of the homology model is dependent on the quality of the sequence alignment and template structure. The approach can be complicated by the presence of alignment gaps (commonly called indels) that indicate a structural region present in the target but not in the template, and by structure gaps in the template that arise from poor resolution in the experimental procedure (usually X-ray crystallography) used to solve the structure. Model quality declines with decreasing sequence identity; a typical model has ~1–2 Å root mean square deviation between the matched Cα atoms at 70% sequence identity but only 2–4 Å agreement at 25% sequence identity. However, the errors are significantly higher in the loop regions, where the amino acid sequences of the target and template proteins may be completely different.Regions of the model that were constructed without a template, usually by loop modeling, are generally much less accurate than the rest of the model. Errors in side chain packing and position also increase with decreasing identity, and variations in these packing configurations have been suggested as a major reason for poor model quality at low identity. Taken together, these various atomic-position errors are significant and impede the use of homology models for purposes that require atomic-resolution data, such as drug design and protein–protein interaction predictions; even the quaternary structure of a protein may be difficult to predict from homology models of its subunit(s). Nevertheless, homology models can be useful in reaching qualitative conclusions about the biochemistry of the query sequence, especially in formulating hypotheses about why certain residues are conserved, which may in turn lead to experiments to test those hypotheses. For example, the spatial arrangement of conserved residues may suggest whether a particular residue is conserved to stabilize the folding, to participate in binding some small molecule, or to foster association with another protein or nucleic acid. Homology modeling can produce high-quality structural models when the target and template are closely related, which has inspired the formation of a structural genomics consortium dedicated to the production of representative experimental structures for all classes of protein folds. The chief inaccuracies in homology modeling, which worsen with lower sequence identity, derive from errors in the initial sequence alignment and from improper template selection. Like other methods of structure prediction, current practice in homology modeling is assessed in a biennial large-scale experiment known as the Critical Assessment of Techniques for Protein Structure Prediction, or CASP.