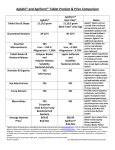
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
KAUNAS UNIVERSITY OF MEDICINE FACULTY OF PHARMACY DEPARTMENT OF DRUGS TECHNOLOGY AND SOCIAL PHARMACY Rasa Kalėdaitė PREPARATION AND DISSOLUTION CHARACTERISTICS OF MATRIX TABLETS BASED ON EUDRAGIT® NM 30 D Master Thesis of Pharmacy Thesis supervisors: Vitalis Briedis, MD PhD, Professor PharmDr. Kateřina Dvořáčková PhD Kaunas 2010 1 SUMMARY The aim of this study was to evaluate the suitability of Eudragit® NM as a matrix forming material for model drugs with different solubility. Methods. Two drugs were selected as the model drugs: freely soluble in water diltiazem hydrochloride and sparingly soluble in water caffeine. Matrix forming agent was chosen Eudragit® NM polymer. Granules obtained by wet granulation were tested according to Ph. Eur. (flowability test, flow test, sieve test and determination of density). Quality parameters of tablets were tested according to Ph. Eur. (mass and content uniformity, hardness, friability and dissolution test). Results. Less than 10 % amount Eudragit® of NM 30 D aqueous dispersion did not ensure the formation of granules, because the amount of binder was not enough. The granulation with more than 30 % amount of aqueous polymer dispersion was made by several steps to avoid forming of wet mass which can not be meshed through the sieve. The flowability time of DH or C granules is small (less than 3,5 s). The flow character of DH and C granules varied from passable to excellent, mostly depending on polymer amount. Higher amount of polymer led to formation of bigger granules and better flow properties. Mass and content uniformity of DH and C tablets were in the limits of Ph. Eur. The friability was very small less than 0,2 %. Hardness of DH and C matrix tablets was more than 100 N, high amounts of polymer led to form a viscous tablets, which deviation of average was high (20 %). Dissolution test was performed 12 hours at pH 6,8. Dissolution profile for 12 hours of DH tablets which contains high amount of MCC and Eudragit® NM was most gradual (4D, 5D) at pH 6.8. Almost all samples of C tablets disintegrated during first hours and did not show almost any retardation of drug release, except 14C sample (25 mg of MCC and 16,68 mg of Eudragit® NM 30 D). Continual dissolution test showed, that the release profile is to fast at pH 1.2, so tablets are recommended to coat acidoresistant coating (Eudragit® L 30 D) 2 SANTRAUKA Tyrimo tikslas yra nustatyti ar Eudragit® NM yra tinkamas naudoti kaip matricą formuojanti medžiaga vaistinėms medžiagoms, pasižyminčiomis skirtingomis fizikocheminėmis savybėmis. Metodai. Vaistų modeliais buvo pasirinktos 2 medžiagos: lengvai tirpus vandenyje diltiazemo hidrochloridas ir ribotai vandenyje tirpus kofeinas. Eudragit® NM polimeras pasirinktas kaip matricą formuojanti medžiaga. Drėgno granuliavimo būdu gautos granulės ištirtos remiantis Europos farmakopėjos metodais (birumo ir takumo testai, sietų analizė bei tankio nustatymas). Tablečių kokybės parametrai buvo ištirti remiantis Europos farmakopėja (masės ir turinio vienodumo testai, tvirtumas, dilumas bei tirpimo testas) . Rezultatai. Mažesnis nei 10 % vandeninės Eudragit® NM 30 D dispersijos kiekis neužtikrino granulių susiformavimo, dėl nepakankamo rišančiosios medžiagos kiekio. Granuliavimas naudojant daugiau nei 30 % vandeninės polimero dispersijos kiekį buvo atliktas keletu etapų, norint išvengti drėgnos masės susiformavimo, kurią sunku pertrinti per sietą. DH ir C granulių birumo laikas buvo mažas (mažiau nei 3,5 s). DH ir C granulių takumo pobūdis kito nuo priimtino iki puikaus, priklausomai nuo polimero kiekio. Didesnis polimero kiekis užtikrino didesnių granulių susiformavimą ir geresnes takumo savybes. DH ir C masės ir turinio vienodumas testų rezultatai buvo Europos Farmakopėjos nustatytose ribose. Dilumas buvo labai mažas (mažiau nei 0,2 %). DH ir C matricos tablečių tvirtumas buvo daugiau nei 100 N, didelis polimero kiekis paskatino susiformuoti atsparias deformacijai tabletes, kurių nuokrypis nuo vidurkio buvo aukštas (20 %). Tirpumo testas buvo atlikinėjamas 12 val pH reikšmė 6.8. Tirpumo profilis DH tablečių, kurios turi didelį kiekį MCC ir Eudragit® NM (4D, 5D) buvo tolygiausias. Beveik visi C tablečių pavyzdžiai suiro per pirmąsias valandas ir beveik neparodė jokio vaisto išsiskyrimo sulėtinimo, išskyrus 14C mėginį (25 mg MCC, 16,68 mg Eudragit® NM). Nuolatinis tirpimo testas parodė, jog tirpumo profilis yra per greitas pH reikšmėje 1,2, taigi rekomenduojama tabletes padengti rūgščiai atsparia danga (Eudragit® L 30 D). 3 ACKNOWLEDMENT I would like to thank Vitalis Briedis, MD PhD, Professor for the opportunity to accomplish research work abroad in University of Veterinary and Pharmaceutical Sciences Brno, Czech Republic. I owe my deepest gratitude to my supervisor, PharmDr. Kateřina Dvořáčková PhD, whose encouragement, guidance and support during research made this thesis possible. I am also grateful to PharmDr. Tereza Bautzová for the assistance during experiments. 4 LIST OF ABBREVIATIONS AGJ - Artificial gastric juice C – Caffeine CSD - Colloidal Silicon Dioxide DH - Diltiazem Hydrochloride Ph. Eur. – European Pharmacopoeia GIT - Gastrointestinal tract MCC - Microcrystalline Cellulose MgS – Magnesium Stearate SD – Standard deviation 5 Table of Contents INTRODUCTION .........................................................................................................................................9 THE AIM AND TASKS OF STUDY ........................................................................................................ 10 1. REVIEW OF LITERATURE ................................................................................................................. 11 1.1. Basic characteristics of Eudragit® types .......................................................................................... 11 1.1.1. pH-dependent types ...................................................................................................................... 11 1.1.1.1. Eudragit®L and S ....................................................................................................................... 12 1.1.1.2. Eudragit® FS .............................................................................................................................. 13 1.1.2. Time-dependent types .................................................................................................................. 13 1.1.2.1. Eudragit® RL and RS................................................................................................................. 14 1.1.2.2. Eudragit® NE ............................................................................................................................. 14 1.1.2.3. Eudragit® NM ............................................................................................................................ 15 1.2. Applications of Eudragit®................................................................................................................ 15 1.2.1. Enteric coatings ............................................................................................................................ 16 1.2.2. Colon and ileum coatings ............................................................................................................. 17 1.2.3. Sustained release .......................................................................................................................... 18 1.2.4. Matrix formulation ....................................................................................................................... 21 1.3. Definition of tablets ......................................................................................................................... 23 1.4. Powder and granules characterization ............................................................................................. 24 1.4.1. Particle size .................................................................................................................................. 24 1.4.1.1. Optical microscopy.................................................................................................................... 25 1.4.1.2. Sieve analysis ............................................................................................................................ 25 1.4.2. Flowing properties........................................................................................................................ 26 1.4.2.1. Flowability ................................................................................................................................ 26 1.4.2.2. Flow (Compressibility index or Hausner ratio) ......................................................................... 27 1.4.3. Measurement of density ............................................................................................................... 29 1.5. Tablets characterization ................................................................................................................... 29 1.5.1. Uniformity of tablets mass ........................................................................................................... 29 1.5.2. Uniformity of tablets content ....................................................................................................... 29 1.5.3. Friability of uncoated tablets ........................................................................................................ 30 6 1.5.4. Resistance to crushing of tablets .................................................................................................. 30 1.5.5. Dissolution test for tablets ............................................................................................................ 31 1.6. Model Drugs .................................................................................................................................... 32 1.6.1. Caffeine ........................................................................................................................................ 32 1.6.2. Diltiazem Hydrochloride .............................................................................................................. 33 1.7. Excipients ........................................................................................................................................ 34 1.7.1. Microcrystalline Cellulose ........................................................................................................... 34 1.7.2. Magnesium Stearate ..................................................................................................................... 34 1.7.3. Colloidal Silicon Dioxide ............................................................................................................. 35 2. EXPERIMENTAL PART ...................................................................................................................... 36 2.1. Drugs and excipients ....................................................................................................................... 36 2.2. Laboratory equipment ..................................................................................................................... 36 2.3. Preparation of granules .................................................................................................................... 37 2.3.1. The measurement of particle size ................................................................................................. 37 2.3.2. Preparation of granules ................................................................................................................. 37 2.4. Evaluation of granules quality parameters ...................................................................................... 39 2.4.1. Determination of granules flowability.......................................................................................... 39 2.4.2. Determination of granules flow (Compressibility index and Hausner ratio) ............................... 39 2.4.3. Determination of density using helium-pycnometer .................................................................... 40 2.4.4. Determination of granules size using sieve analysis .................................................................... 40 2.5. Preparation of matrix tablets ........................................................................................................... 40 2.5.1. Preparation of granules for compressing ...................................................................................... 40 2.5.2. Compression of matrix tablets ...................................................................................................... 41 2.6. Evaluation of quality parameters of matrix tablets (Ph. Eur.) ......................................................... 42 2.6.1. Uniformity of tablet mass ............................................................................................................. 42 2.6.2. Uniformity of tablet content ......................................................................................................... 42 2.6.3. Friability of tablets ....................................................................................................................... 43 2.6.4. Resistance to crushing of tablets (Hardness of tablets) ................................................................ 43 2.6.5. Determination of released drug from matrix tablet ...................................................................... 43 3. RESULTS AND DISCUSSION ............................................................................................................ 45 7 3.1. Preparation of granules .................................................................................................................... 45 3.2. Results of granules evaluation ......................................................................................................... 46 3.3. Results of tablets evaluation ............................................................................................................ 47 CONCLUSIONS ........................................................................................................................................ 49 REFERENCES ........................................................................................................................................... 50 ADDITION Nr. 1 ....................................................................................................................................... 55 ADDITION Nr. 2 ....................................................................................................................................... 58 ADDITION Nr. 3 ....................................................................................................................................... 66 ADDITION Nr. 4 ....................................................................................................................................... 70 8 INTRODUCTION Eudragit® is the product line which includes pharmaceutical copolymers from esters of acrylic or methacrylic acid whose properties are determined by functional group. The individual grades differ in their proportion of neutral, alkaline or acid groups and thus in terms of their physicochemical properties. Depending on the pH, these polymers act as polyelectrolytes which make them suitable for different purposes, from gastric or intestinal soluble drug formulations to insoluble but swellable delivery forms (matrix formulations), regulated by percentage of charged and nonionized (ether) groups in the structure of these copolymers. [2] Anionic Eudragit® L, S and FS types dissolve in neutral or alkaline fluids. Insoluble Eudragit® RL/RS types have hydrophilic quaternary ammonium groups as hydrochlorides, providing different permeability, whereas the insoluble Eudragit® NE/NM types include no functional groups. These insoluble polymers absorb water from physiological fluids and swell in a pH-independent way to create diffusional barriers for time-controlled drug release. [1] In order to control chronic diseases (arterial hypertension, ischemic heart disease, arthritis) is important to maintain permanent drug concentration in blood or tissues, sustained release drug forms are suitable to solve this problem. Eudragit® NM is a new time-dependent polymethacrylate polymer, which is suitable for sustained release formulations. Reviewing of the various sources of scientific literature found that there is little information about Eudragit® NM. There are done just little researches with water dispersion of Eudragit® NM (30% water dispersion) a matrix forming agent, so it was topical to make a research with Eudragit® NM as the drug release modifier. 9 THE AIM AND TASKS OF STUDY The aim of study is to evaluate Eudragit® NM suitability as a matrix forming agent for model drugs with different solubility. Tasks of study: 1. To prepare granules from two materials with different solubility (DH and C), using different quantity of MCC and Eudragit® NM 30 D by wet granulation method. 2. To evaluate of prepared DH and C granules physical properties and suitability for tablets manufacturing by Ph. Eur. 3. To press matrix tablets of DH and C and to assess their quality parameters by Ph. Eur. 4. To determine the most appropriate amount of MCC and Eudragit® NM in matrix tablets of DH and C which provides prolonged drug release for 12 hours at pH 6,8. 10 1. REVIEW OF LITERATURE 1.1. Basic characteristics of Eudragit® types 1.1.1. pH-dependent types The main structural element of the synthetic methacrylate copolymers is an acidic function (phthalate or methacrylic acid), which is responsible for the pH-dependent dissolution. [15] The carboxylic groups are transformed to carboxylate groups in the pH range of 5 – 7 by salt formation with alkali or amines. Their dissolution pH depends primarily on their content of carboxylic groups. Methacrylic acid – methyl methacrylate copolymer 1:1 (Eudragit® L 100) dissolves at pH 6, methacrylic acid – methyl methacrylate copolymer 1:2 (Eudragit® S 100) – above pH 7. When the ester component is more hydrophilic ethyl acrylate, the films prepared from methacrylic acid – ethyl acrylate copolymer 1:1 (Eudragit® L 30D or redispersed powder L 100-55) dissolves above pH 5.5. [1] Table 1. pH – dependent Eudragits® [3, 16, 18] Eudragit® Polymer Availability L 30 D-55 30 % Aqueous Dispersion L 100-55 Powder L 100 Powder L 12,5 12,5 % Organic Solution S 100 Powder S 12,5 12,5 % Organic Solution FS 30 D 30 % Aqueous Dispersion Dissolution Properties Dissolution above pH 5,5 Dissolution above pH 6,0 Dissolution above pH 7,0 11 1.1.1.1. Eudragit®L and S Figure 1. Eudragit® L 30 D-55 [4] Eudragit® L 30 D-55 (methacrylic acid – ethyl acrylate copolymer 1:1 dispersion 30 per cent Ph. Eur.) is the aqueous dispersion with 30% of dry substance of an anionic copolymer based on methacrylic acid and ethyl acetate. The dispersion contains 0.7 % sodium laurilsulfate and 2.3 % polysorbate 80 on solid substance, as emulsifiers. The ratio of the free carboxyl groups to the ester groups is approx. 1:1. A molecular mass is about 250 000. [4, 17] Eudragit® 100-55 (methacrylic acid – ethyl acrylate copolymer 1:1, type A Ph. Eur.) is a solid substance. The product contains 0.7 % sodium laurilsulfate and 2.3 % polysorbate 80 on solid substance, as emulsifiers. [5] Figure 2. Eudragit® L, resp. Eudragit® S [6] Eudragit® L 100 (methacrylic acid – methyl methacrylate copolymer 1:1 Ph. Eur.) and Eudragit® S 100 (methacrylic acid – methyl methacrylate copolymer 1:2 Ph. Eur.) are solid substances. The ratio of free carboxyl groups to the esters is about 1:1 in Eudragit® L 100 and 1:2 in Eudragit® S 100. The relative molecular mass is about 135 000. [6, 17] Eudragit® L 12,5 (methacrylic acid – methyl methacrylate copolymer 1:1 Ph. Eur.) and Eudragit® S 12,5 (methacrylic acid – methyl methacrylate copolymer 1:2 Ph. Eur.) is a solution of Eudragit® L 100 (Eudragit® S 100 Ph. Eur.) with 12.5 % dry substance in aqueous isopropyl alcohol. [7] 12 1.1.1.2. Eudragit® FS Figure 3. Eudragit® FS. [8] Eudragit® FS 30 D is the aqueous dispersion of an anionic copolymer based on methyl acrylate, methyl methacrylate and methacrylic acid. The dispersion contains 0.3 % sodium laurilsulfate and 1.2 % polysorbate 80 on solid substances, as emulsifiers. The ratio of the free carboxyl groups to the ester groups is 1:10. The average molecular weight is about 220 000. [8] 1.1.2. Time-dependent types After contact with gastrointestinal fluids, the film coatings swell, independently of pH, and release the active substance by a diffusion-controlled mechanism. [1] Table 2. Time-dependent Eudragits® [3,22] Eudragit® Polymer Availability Dissolution Properties RL 100 Granules RL PO Powder RL 30 D 30 % Aqueous Dispersion RL 12,5 12,5 % Organic Solution RS 100 Granules RS PO Powder Low permeability RS 30 D 30 % Aqueous Dispersion pH-independent swelling RS 12,5 12,5 % Organic Solution NE 30 D 30 % Aqueous Dispersion Insoluble, low permeability NE 40 D 40 % Aqueous Dispersion pH-independent swelling NM 30 D 30 % Aqueous Dispersion No plasticizer required Insoluble High permeability pH-independent swelling Insoluble Highly flexible 13 1.1.2.1. Eudragit® RL and RS Figure 4. Eudragit® RL and RS. [9] Eudragit® RL 100 (ammonio methacrylate copolymer type A Ph. Eur.) and Eudragit® RS 100 (ammonio methacrylate copolymer type B Ph. Eur.) are solid substances; Eudragit® RL PO and Eudragit® RS PO are solid substances obtained from Eudragit® RL 100 or Eudragit® RS 100. They are copolymers of ethyl acrylate, methyl methacrylate and low content of methacrylic acid ester quaternary ammonium groups (trimethylammonioethyl chloride). The ammonium groups are present as salts and make the polymers permeable. The average molecular weight is 150 00. [9] Eudragit® RL 30 D and Eudragit® RS 30 D are aqueous dispersions of Eudragit® RL 100 or Eudragit® RS 100 with 30 % dry substance. The dispersions contain 0.25 % sorbic acid as a preservative and 0.1 % sodium hydroxide as an alkalizing agent. [10] Eudragit® RL 12,5 and Eudragit® RS 12,5 are solutions of Eudragit® RL 100 or Eudragit® RS 100 with 12.5 % (w/w) dry substance in a mixture of 60 % (w/w) isopropyl alcohol Ph. Eur. and 40 % (w/w) acetone. [11] 1.1.2.2. Eudragit® NE Figure 5. Eudragit® NE. [12] Eudragit® NE 30 D (polyacrylate dispersion 30 per cent Ph. Eur.) is the aqueous dispersion with 30 % dry substance of a neutral copolymer based on ethyl acrylate and methyl methacrylate. The dispersion contains 1.5 % nonoxynol as an emulsifier. The average molecular weight is 800 000. [12] 14 Eudragit® NE 40 D is identical to Eudragit® NE 30 D with 40 % dry substance instead of 30 % dry substance. The dispersion contains 2.0 % nonoxynol as an emulsifier. [13] 1.1.2.3. Eudragit® NM Figure 6. Eudragit® NM. [14] Eudragit® NM 30 D (polyacrylate dispersion 30 per cent Ph. Eur.) is an aqueous dispersion with 30 % dry substance of a neutral copolymer based on ethyl acrylate and methyl methacrylate. The dispersion contains 0.7 % macrogol stearyl ether Ph. Eur. as an emulsifier. The aqueous dispersion is miscible with water in any proportion, the milky-white appearance being retained. When 1 part NM 30 D is mixed with 5 parts acetone, a clear to slightly cloudy, viscous solution is obtained. The same occurs when mixed with ethanol or isopropyl alcohol; initially the polymer is precipitated, but then dissolves again in the excess organic solvent. When mixed with 1 N sodium hydroxide in a ratio of 1:2, the dispersion does not dissolve. The milkywhite appearance is retained. The average molecular weight is 600 000. [14] 1.2. Applications of Eudragit® Generally in technology of solid dosage forms, Eudragits® can be used as the coating materials (coated tablets, capsules, pellets, microparticles) and matrix formers (matrix tablets, pellets, microparticles). As the coating materials Eudragits® are able to ensure on one hand site specific delivery of active ingredient (enteric, ileic, colonic) because of their pH-dependent solubility and on the other hand pH-independent types are of use in sustained drug release. The other Eudragits® application is their incorporation into matrix systems where effectively slow down the drug release in time. 15 Figure 7. Eudragit® polymers used for oral solid dosage formulations. [35] 1.2.1. Enteric coatings Enteric-coated formulations are suitable to modify the release of the active substance that it would be released at the proximal part of small intestine. The intended use includes drug stabilization within the stomach passage; protection against stomach irritation and release directed to defined segments in the digestive tract. [15] The small intestine can be targeted with polymers having solubility at pH in the range between 5.0 – 6.0, the distal part requires polymers having solubility at pH in the range between 7.0 – 7.5. The pH sensitive material is insoluble or almost impermeable in dissolution liquids of low pH but can dissolve in those liquids which pH is from 5 to 7. The approach depends on the GIT transit time, which differs in individuals. [20] Polymers can be used also to create the drug form with pulsatile release. Fan et al. investigated the pulsatile release tablets with ethylcellulose and Eudragit® L as film coating materials. The purpose of study was to develop new pulsatile release tablets, which can suppress drug release in stomach and release drug rapidly after a lag time period in intestine. Dissolution of Eudragit® L causes pores in the film, so it was selected as the coating material for this purpose. The lag time 16 can be controlled by the thickness of the coating film. Water penetrates through the coating film and causes the water uptake and expansion of swelling material until the internal forces on the film causes the tablet to burst and for that reason the drug is released. The burst of thicker film needs more powerful forces and leads to longer lag time than thinner film. [19] 1.2.2. Colon and ileum coatings Colon-specific targeting is used for the topical treatment of local disorders. [18] The second approach is to use a material which dissolves above pH 7. It is suitable for the colonic delivery system and for this purpose are used Eudragit® S and FS. Many commercial drug formulations for the oral treatment of inflammatory diseases (such as Asacolitin®, Claversal®, Salofalk® or Budenofalk®) are coated with pH-sensitive colon coating polymers such as Eudragit® L or S® (see Table 3) [38]. The solubility in pH 7 can limit the drug release in the proximal part of the GIT, but there is a possibility that no drug will be released in the colon if the film is too thick. The release rate of drug coated with Eudragit® S100 depends not only the pH but also on the ion concentrations of the in vitro solutions. The faster release of the drug is due to to a higher concentration of ions, because the carboxylic groups in the Eudragit ® S100 are reacting with bases in the liquid and it causes the increased film solubility rate. [20] Ibekwe et al. were investigating the in vitro dissolution characteristics of pH-dependent poly(meth)acrylate polymers in a different simulated fluids. Tablets coated with Eudragit ® S aqueous dispersion showed a faster dissolution rate comparing to Eudragit® S organic solution, because of the partial neutralization of the methacrylic acid units which are responsible for the pH-dependent solubility, during the re-dispersion process. The dissolution of Eudragit® S aqueous coated tablets begins in the proximal part of the ileo-colonic region. Low permeability of formulations containing Eudragit FS to water vapor is due to a low hydrophilic character of this polymer. Tablets coated with Eudragit® FS are suited for delivery to the ileo-colonic region, but polymer was observed to exhibit a pH-dependent permeability to aqueous media, with some degree of moisture uptake across the entire pH range used in the dissolution tests (6.8 – 7.4) and swelling around the tablet core prior to presumable drug release at pH more than 7. [21, 23] 17 Table 3. Coated dosage forms for the treatment of ulcerative colitis in the German market. [38] Drug Coating polymers ® Dissolution pH Trade name ® Mesalazine Eudragit L 6.0 Claversal Mesalazine Eudragit® S 7.0 Asacolitin® Mesalazine Eudragit® L 6.0 Salofalk® Sulfasalazine Eudragit® L 100-55 5.5 Colo-Pleon® Budenoside Eudragit® L 100- 5.5 55, ethylcellulose Eudragit® S 6.0 Budenoside Entocort® Budenofalk® Manufacturer SmithKline Beecham Pharmaceuticals Munich Henning Berlin GmbH & Co., Berlin Dr Falk Pharma GmbH, Freiburg Henning Berlin GmbH & Co., Berlin ASTRA GmbH, Wedel Dr Falk Pharma GmbH, Freiburg 1.2.3. Sustained release Sustained release polymeric film coating is based upon a generic reservoir design in which the release of a concentrated drug core is controlled by a semi-permeable membrane. The membrane controls the fluid permeation into the drug core, thereby controlling the dissolution and subsequent outward diffusion of the active substance. The primary benefit of sustained release dosage forms is the reduction of the daily dosing to a twice or once-daily schedule. By reducing of the required daily doses number, the drug therapy is improved by better patient compliance and often reduced cost. A sustained release dosage form can stabilize the systemic drug concentration by providing a constant rate of drug release and absorption. This is very important for certain applications, for example a pain therapy, that patient could rest throughout the night without suffering the pain. [22] There are some drugs, which have a narrow therapeutic index, for example theophylline. It is a xanthine derivative, which is used in the treatment of bronchial asthma and bronchospastic diseases. So, it requires suitable formulation to maintain the drug concentration in the serum within the therapeutic range and sustained-release oral formulation is the best solution. [27] The polymethacrylates that are used for sustained-release film coatings are Eudragit® RL (highly permeable), Eudragit® RS (low permeable), Eudragit® NE (permeable), Eudragit® NM (permeable). These systems are composed of polymers that are water insoluble, but swellable over the range of physiological pH, and they are suitable for sustained release film coating applications. [1, 21] Eudragit RL and RS have quaternary ammonium groups which are in the chloride salt form. The dissociation of these groups in aqueous media is responsible for the hydration and swelling of the polymers films. [22] Eudragit® RL 100 includes a greater 18 concentration of quaternary ammonium groups and the coatings made from this polymer are more permeable than those which are made from Eudragit® RS 100. So, the drug release through Eudragit® RL 100 membranes is bigger than from Eudragit® RS 100. The sustained-release Eudragit® polymer are used of coating materials for pellets [22], tablets [19], capsules [20], microspheres [22]. The drug release from dosage forms coated by these polymers could be modified by addition of wide range other excipients. To increase the permeability of film layers such substances can be added like: sucrose, lactose and other saccharides; starch, micronized cellulose, soluble cellulose ethers; poly(vinylpyrrolidone), polyethylene glycol or its derivatives and fumed or precipitated silica. But water-soluble cellulose ethers have limited compatibility, because they stimulate slow agglomeration and coagulation within few hours or days [1]. For instance, the coating permeability from low permeable Eudragit® RS can be increased with addition of inulin to the film. Inulin is a naturally occurring polysaccharide which is not significantly hydrolyzed by gastric or intestinal enzymes. The increased swelling ratio is due to the presence of inulinase enzyme in the dissolution media (simulated colonic fluid) which can diffuse into the polymeric chains, hydrolyze the fructose backbone of inulin, reduce the network density and increase the swelling ratio. Colonic bacteria (specifically Bifidobacteria) can degrade this polysaccharide and this increases the permeability of the film [23]. Eudragit® RL 100 and RS 100 coating systems have been used in a different sustained release coating applications. [1, 21, 23]. Often, Eudragit® RS and (or) RL are used like coating materials to create sustained release drug forms from such active substances like ibuprofen, indomethacin, nitrendipine, diltiazem and others [22]. Eudragit® RL and RS could be combined with other Eudragit® polymer to achieve the desirable dissolution profile. Eudragit® RL 30 D and RL 30 D in combination with Eudragit® FS 30 D were used as coating materials to produce sustained release pellets of 5-aminosalicylic acid (5-ASA) for the colon targeting. Pellets were prepared by powder layering of 5-ASA on nonpareils (0.5-0.6 mm) in a conventional coating pan. Then pellets were coated with an inner layer of Eudragit® RL and RS (8:2) and a outer layer of Eudragit® FS (different amounts). The release profile of 5-ASA was sustained for more than 12 h in phosphate buffer after simulated gastric pre-soak for 2 h. (see Figure 8). [22, 37] 19 Figure 8. Dissolution of 5-ASA pellets for the first 2 h at pH 1.2 followed by pH 7.0 phosphate buffer. [37] Different ratios of Eudragit® NE 30 D and Eudragit® L 30D-55 were tested as the coating materials for drug-layered beads. These were prepared by spraying of Eudragit ® RS 30D dispersion containing verapamil-hydrochloride as the model drug. It was found that suitable ratios of polymers were 75:25 and 80:20. In 60% coating level these combinations were suitable as a functional film coating material for use in delayed drug release applications. For 75:25 polymer ratio the lag time was approx. 3 hours. The longer lag time approx. 5 hours was observed in the case of 80:20 polymer one. Generally, the lag time increased and drug release decreased with increasing amount of Eudragit® NE 30 D in the polymer blend (see Figure 9). [24] 20 Figure 9. Dissolution profiles of uncoated beads and beads coated with NE 30D, L30D and blends thereof at ratios 75:25 and 80:20 at 60% weight gain. [24] 1.2.4. Matrix formulation Matrices are monolithic systems constituted of active substance dispersed and entrapped in a continuum of excipient (adjuvant) – the “matrix forming” substance. The special matrix advantage is the non-immediate disintegration of the monolith in contact with a dissolution liquid. The other advantages are simplicity of preparation and low product costs. The usual appearance of the matrix is the tablet. The matrix keeps a structure for the time needed to release the dispersed or dissolved drug. The dissolution is slowed down by the typical release mechanism. There are three types of matrices, which can be constructed and their release kinetics changing according to the category – inert, erodible and swellable matrices. Inert matrices leave residual skeletons, erodible matrices slowly disintegrate and the swellable matrices forms gel layer. [25] The swellable matrix undergoes erosion during its release time, but the drug release can proceed together or in the different time with the matrix erosion or dissolution. It depends on the combination of hydrophilic polymers used for the making the matrix. The swellable matrices are typical moving boundary release systems. The drug release is controlled by continuously changing dimension of the diffusive barrier. This barrier is the layer thickness externally formed on the matrix that controls active substance transport through it. In swellable matrices the barrier is called gel layer. The hydrophilic polymer fraction in the matrix is the most important parameter for determining drug release profile. Drug solubility is important also for the release kinetics. Highly soluble drugs act as pore formers leading to a fast drug release by diffusion 21 process. Poorly soluble drugs will be released mainly by matrix erosion. Many drugs are released from matrix in the combination of two processes – the diffusion and the erosion. [25] The whole process consists from: diffusion of the liquid through the Eudragit® polymer matrix and into branched polymer, reaction between the branched polymer and the liquid, dissolution of the drug in the liquid, diffusion of the drug through the liquid located in the branched polymer and the polymer matrix. [26] Matrices are easily manufactured by direct compression and compression of granules which are obtained by dry, wet (high shear mixer or fluidized bed) or melt granulation. [28] Eudragit® is attractive like a matrix forming materials, due to their high chemical stability, good compatibility properties and large variety of products with different physicochemical characteristics present on the market. A swellable matrix can be formed from some Eudragit ® polymers. Ceballos et al prepared extended-release theophylline matrix tablets by a direct compression of drug and different pH-dependent (Eudragit® L 100, S 100 AND L 100-55) and time-dependent (Eudragit® RL PO and RS PO) polymer combinations. Matrix tablets based on L 100/RL PO and L 100/RS PO mixtures gave the best results, displaying the highest percent of theophylline released and the matrix formulation allowed to obtain the more regular release profiles, with the best equilibrium between the values of drug releases amount at the gastric and intestinal pH. This was made by the combination of the good erodible properties of L100 with the swelling properties of RLPO and RSPO polymers. The use of a mixture of Eudragit L100 and RLPO in the 0.7:0.3 w/w ratio enabled a highly reproducible drug release profile to be achieved, with an almost zero-order kinetic. [27] Colo et al. reported that compressed matrix tablets based on pH-sensitive polyethylene oxide and Eudragit® L100 compounds ensure a complete release of the active substance during the transit from stomach to jejunum. A ionization of the Eudragit® L carboxyls by the anions is the main step of the release from the matrices to the intestinal fluids. [16] Inert matrix tablets of carteolol hydrochloride can be prepared from Eudragit ® RS as a supporting material with different fillers and wetting liquids. Carteolol has a potent β-adrenergic blocking action. Use of lactose does not allow to form an inert matrix, because this formulation shows a disintegration process depending on its hydrophilic nature. Mannitol, polyethylene glycol 6000 and Emcompress® (calcium hydrogen phosphate dihydrate and dibasic calcium phosphate) are suitable as fillers. The use of Eudragit ® L12.5 as a wetting liquid allows to obtain two phase release profile. The first phase may represent the release of a drug on the surface of a tablet and the particles of the drug which are not completely surrounded by the Eudragit ®. The second phase is the release of drug contained in a inert matrix. [29] Shanawany was investigating 22 sustained release granules of nitrofurantoin from inert wax matrices. Nitrofurantoin is used to treat urinary tract infection. The problem is that the drug did not achieve therapeutical concentration because it rapidly eliminated. The long term treatment produces side effect in the gastrointestinal tract. In order to minimize the side effect and to maintain the blood level within therapeutic range it was decided to formulate the drug as a sustained release preparation using an inert wax matrix. The release of a drug from an inert wax matrix involves leaching by the dissolution media that contacts the embedded drug. The fluid can enter the core through pore channels and cracks. Several materials have been used as channeling or pore forming agents to improve the release of the drug from wax matrices: colloidal silicone dioxide, microcrystalline cellulose, dibasic calcium phosphate hydrate. The release of the drug was significantly increased with increase of channeling agents. The granules were prepared by fusion, solvent evaporation or melt granulation methods. Granules prepared by the fusion method and containing equal quantities of stearic acid and glyceryl monostearate showed the best sustained release properties. [28] In erodible matrix systems the mechanism of the drug release occurs by erosion. The difference of these systems from inert matrices is that the polymer in the inert systems remains unchanged with time and the drug is released by diffusion and the polymer phase in erodible systems decreases with time. The erosion mode of these delivery systems is one of the factors controlling drug release. There are two different modes of erosion: surface (heterogeneous) and bulk (homogeneous). In bulk-degrading systems, degradation occurs homogeneously throughout the bulk of the system. In surface-degrading systems, degradation is confined to the outer surface of the system. The rate of the drug release from a surface-eroding device is proportional to the surface area of the delivery system. [30] An erodible matrix can be made from hydroxypropylmethylcellulose. Karasulu et al reported that even a geometrical shape of the tablets affect the release rate of the active substance (theophylline) in erodible hydrogel matrix system. Three geometrical shapes were investigated. The highest release rate had triangular tablets, then – half-spherical and the lowest rate had cylindrical tablets. [31] 1.3. Definition of tablets Tablets are solid preparations each of which contains a single dose of one or more active substance. They are obtained by compressing uniform volumes of particles, and they are almost always intended for oral administration. Compressing pharmaceutical tablets is the most efficient process for producing a single dose for medication. [32] Some are swallowed whole, some after 23 being chewed, some are dissolved or dispersed in water before being administered and some are retained in the mouth where the active substance is liberated. The particles consist of one or more active substances with or without excipients such as diluents, binders, disintegrating agents, glidants, lubricants, substances capable of modifying the behavior of the preparation in the digestive tract, coloring matter authorized by the competent authority and flavoring substances. Tablets are usually right, circular solid cylinders, the end surfaces of which are flat or convex and the edges of which may be beveled. They may have lines or break-marks and may bear a symbol or other markings. Tablets may be coated. [17] Tablet drug delivery systems can range from relatively simple immediate-release formulations to complex extended- or modifiedrelease dosage forms. The most important role of tablet is to achieve the drug delivery to the site of action in sufficient amount and at the appropriate rate, but it must also meet a number of other essential criteria. These include physical and chemical stability, ability to be economically mass produced in a manner that assures the proper amount of drug in each and every dosage unit and in each batch produced, and, as far as possible, patient acceptability (reasonable size and shape, taste, color, etc. to encourage patients to take the drug and thus comply with the prescribed dosing regimen). [33] The compressed tablet is by far most widely used dosage form, because they are easily administrated and simple to use. The tablet is the most popular dosage form because it provides advantages for all concerned in the production and consumption of medicinal products. For the manufacturer it is considerable, because the tablets can be produced at a much higher rate than any other dosage form. The tablet is a dry dosage, so it promotes stability and they have a long shelf lives measured in years. Tablets are also convenient to transport in bulk. From the viewpoint of the pharmacist, tablets are easy to dispense, while the patient receives a concentrated and readily consumed dosage form. The appropriate coating can mask unpleasant tastes and improve patient acceptance. [32] 1.4. Powder and granules characterization 1.4.1. Particle size A powder is characterized by its particle size, which is important to achieve optimum qualities of tablets. This parameter influences the dissolution rate of the drug in vivo, which in turn influences absorption rate and therapeutic activity. Particle size is important during the production of solid dosage forms like tablets and capsules. The manufacture of tablets is based by a volumetric method. Powders with different particle sizes have different flow and packing properties, which influences the volume of powders during manufacture process. Any 24 interference with the uniformity of fill volumes may alter the mass of the drug incorporated into the tablet and reduce the content uniformity. To avoid such problems, the particle size should be defined during formulation and must be as uniform as possible. [33] There are many different methods available for particle size analysis. The most common techniques used in tablet production and raw material processing include sieving, optical microscopy in conjunction with image analysis, electron microscopy, laser diffractometers and etc. The particle size measurement method depends on the approximate particle size range. [36] 1.4.1.1. Optical microscopy Direct measurement of particle dimensions is possible from enlarged photographic or electronic images of microscopes. There are three types of microscopes commonly used – the optical microscope, the scanning electron microscope and the transmission electron microscope. The optical microscope is used to measure particles from 1 µm to about 150 µm. The other microscopes make use of electron beams and can be used for particles 0.01 µm to 5 µm. They are especially useful for revealing the surface morphology of extremely small particles. Particles to be imaged in an optical microscope are usually dispersed in a drop of viscous fluid in which they are not soluble on a glass slide. [40] A solid particle is often characterized by a diameter. The measurement is based on a hypothetical sphere that represents only an approximation to the true shape of the particle. [33] The microscopic measurement technique is most suitable for particles relatively uniform in size and granular in shape, because a large number of particles, between 300 to 500, need to be measured to minimize statistical error. [40] 1.4.1.2. Sieve analysis The most commonly used method for classifying powders (especially granules) is to sieve the particles through a series of screens with standardized mesh size by sifting, swirling, shaking or vibrating. Sieve analysis does not provide the information for the largest and the smallest particle sizes. This analysis also does not differentiate the particle shape. The result of sieve analysis is also dependent on the time of sieving action, the particle loading on the sieve and sieve blinding. Method. A weighed sample is poured into the top sieve which has the largest screen meshes. Each lower sieve has smaller meshes than the above one. At the base is a round pan, called receiver. A sample is shaken for a fixed time period at a given amplitude and pulse frequency. After the shaking the material on each sieve is weighed. 25 Results. The weight of powder on each sieve can then be calculated and the particle size distribution obtained. A mean sieved diameter is calculated. Because the weight of particles on each sieve is determined, the mean sieved diameter represents a mass distribution. The results are expressed by the percentage of each portion from the total amount. The sieve analysis gives a result of an approximate value for the mean particle size. [40, 41] 1.4.2. Flowing properties During many pharmaceutical production processes, it is necessary to transfer large quantities of powder from one location to another in a controlled manner. For example, in powder blending, powder filling into the dies of a tablet press, powder flow into capsules and etc. For this reason, the powders for pharmaceuticals use must have sufficient properties of flowing. [41] The fluidity of powder is influenced by various properties of the particles, such as particle size and its distribution, shape and surface roughness of the particles, moisture and the interparticle forces. The fluidity of a powder can be improved by changing its physical properties, such as moisture content and particle size and shape, by drying, grinding, classification and granulation. [33] 1.4.2.1. Flowability Flowability is the ability of powder (granules) to flow in a desired manner is a specific piece of equipment. [41] The test for flowability is intended to determine the ability of divided solids (for example, powders and granules) to flow vertically under defined conditions. Apparatus. According to the flow properties of the material to be tested, funnels with or without stem, with different angles and orifice diameters are used (see Table 4). Typical apparatuses are shown in Figure 10. The funnel is maintained upright by a suitable device. The assembly must be protected from vibrations. Method. Into a dry funnel, whose bottom opening has been blocked by suitable means, introduce without compacting a test sample weighed with 0.5 per cent accuracy. The amount of the sample depends on the apparent volume and the apparatus used. Unblock the bottom opening of the funnel and measure the time needed for the entire sample to flow out of the funnel. Carry out three determinations. Results. The flowability is expressed in seconds and tenths of seconds, related to 100 g of sample. [17] 26 Figure 10. Typical apparatus for the test of powder (granules) flowability. [17] Table 4. The sizes of nozzles, which can be used for the flowability test. [17] Nozzle Diameter of the outflow opening (mm) 1 10 ± 0.01 2 15 ± 0.01 3 25 ± 0.01 1.4.2.2. Flow (Compressibility index or Hausner ratio) The widespread use of powders in the pharmaceutical industry has generated a variety of methods for characterizing powder flow. Four commonly reported methods for testing powder flow are: 1. Angle of repose, 2. Compressibility index or Hausner ratio, 3. Flow rate through an orifice, 4. Shear cell. The development of such a variety of test methods was inevitable; powder behavior is multifaceted and thus complicates the effort to characterize powder flow. [17] In recent years the compressibility index and the closely related Hausner ratio have become the simple, fast, and popular methods of predicting powder flow characteristics. The compressibility index has been proposed as an indirect measure of bulk density, size and shape, surface area, moisture content, and cohesiveness of materials, because all of these can influence the observed compressibility 27 index. [17] Bulk or tapped density is a measure of the degree of packing or, conversely, the amount of space between the particles in the powder. Bulk density is determined by placing a sample of powder (granules) of known weight in a graduated cylinder. Tapped density is determined by tapping the powder in the graduated cylinder until it no longer settles. [41] The compressibility index and the Hausner ratio are determined by measuring both the bulk volume and tapped volume of a powder. Method. The basic procedure is to measure the unsettled apparent volume, (V0), and the final tapped volume, (Vf), of the powder after tapping the material until no further volume changes occur. The bulk and tapped densities; compressibility index and the Hausner ratio are calculated as follows: (1) (2) (3) (4) Results. For the compressibility index and the Hausner ratio, the generally accepted scale of flowability is given in Table 5. [17] Table 5. Scale of powder (granules) flowability. [17] Compressibility index (%) Flow character Hausner ratio 1-10 Excellent 1.00-1.11 1-15 Good 1.12-1.18 16-20 Fair 1.19-1.25 21-25 Passable 1.26-1.34 26-31 Poor 1.35-1.45 32-37 Very poor 1.46-1.59 > 38 Very, very poor > 1.60 28 1.4.3. Measurement of density Density is mass per unit volume; it is expressed in grams per cm3. Density can be measured by pycnometer. Pycnometers are used for research and quality control in such industries as ceramics, fibers, minerals, pharmaceuticals and others. Helium-pycnometry is a technique to obtain information on the true density of solids. Since helium, which can enter even the smallest voids or pores and is the least adsorptive, is used to measure, the final result is often referred to as skeletal density. [45] Method. Turn on the helium pycnometer and open helium gas faucet. Wait till temperature will be 20° C. Weigh a dry clean metal vessel. Add approximately 10 g of powder (granules). Weigh a vessel with material again. Put the vessel with material into a measurement place and close it. Enter the values of weight of empty vessel and loaded vessel. Start the measurement. Results. It is expressed in grams per cm3. 1.5. Tablets characterization 1.5.1. Uniformity of tablets mass Weigh individually 20 tablets taken at random and determine the average mass. Not more than 2 of the individual masses deviate from the average mass by more than the percentage deviation shown in Table 6 and none deviates by more than twice that percentage. Table 6. Tablet deviation of mass. [17] Pharmaceutical form Average Mass Percentage Deviation Tablets (uncoated and 80 mg or less 10 film-coated) More than 80 mg and less than 250 mg 7.5 250 mg or more 5 1.5.2. Uniformity of tablets content The test for uniformity of content of tablets is based on the assay of the individual contents of active substance(s) of a number of single-dose units to determine whether the individual contents are within limits set with reference to the average content of the sample. 29 Method. Using a suitable analytical method, determine the individual contents of active substance(s) of 10 tablets taken at random. Results. The preparation complies with the test if each individual content is between 85 % and 115 % of the average content. The preparation fails to comply with the test if more than one individual content is outside these limits or if one individual content is outside the limits of 75 % to 125 % of the average content. If one individual content is outside the limits of 85 % to 115 % but within the limits of 75 % to 125 %, determine the individual contents of another 20 tablets taken at random. The preparation complies with the test if not more than one of the individual contents of the 30 units is outside 85 % to 115 % of the average content and none is outside the limits of 75 % to 125 % of the average content. 1.5.3. Friability of uncoated tablets This test is for the friability determination of compressed, uncoated tablets. Measurement of tablet friability supplements other physical strength measurements, such as tablet breaking force. Method. Use a drum, with an internal diameter between 283-291 mm and a depth between 36-40 mm, of transparent synthetic polymer with polished internal surfaces, and subject to minimum static build-up (see Figure 16). For tablets with a unit mass equal to or less than 650 mg, take a sample of whole tablets corresponding as near as possible to 6.5 g. The tablets are carefully dedusted prior to testing. Accurately weigh the tablet sample, and place the tablets in the drum. Rotate the drum 100 times, and remove the tablets. Remove any loose dust from the tablets as before, and accurately weigh. Generally, the test is run once. If obviously cracked, cleaved, or broken tablets are present in the tablet sample after tumbling, the sample fails the test. If the results are difficult to interpret or if the weight loss is greater than the targeted value, the test is repeated twice and the mean of the 3 tests determined. Results. A maximum loss of mass (obtained from a single test or from the mean of 3 tests) not greater than 1.0 % is considered acceptable for most products. [17] 1.5.4. Resistance to crushing of tablets This test is intended to determine, under defined conditions, the resistance to crushing of tablets, measured by the force needed to disrupt them by crushing. Apparatus. The apparatus consists of 2 jaws facing each other, one of which moves towards the other. The flat surfaces of the jaws are perpendicular to the direction of movement. 30 The crushing surfaces of the jaws are flat and larger than the zone of contact with the tablet. The apparatus is calibrated using a system with a precision of 1 Newton. Method. Place the tablet between the jaws, taking into account, where applicable, the shape, the break-mark and the inscription; for each measurement orient the tablet in the same way with respect to the direction of application of the force. Carry out the measurement on 10 tablets, taking care that all fragments of tablets have been removed before each determination. Expression of results. Express the results as the mean, minimum and maximum values of the forces measured, all expressed in Newton’s. [17] 1.5.5. Dissolution test for tablets The test is used to determine the dissolution rate of the active ingredients of tablets. Paddle apparatus consists of: 1. a cylindrical vessel of borosilicate glass or other suitable transparent material with a hemispherical bottom and a nominal capacity of 1000 ml ; a cover is fitted to retard evaporation; the cover has a central hole to accommodate the shaft of the stirrer and other holes for the thermometer and the devices used to withdraw liquid; 2. a stirrer consisting of a vertical shaft to the lower end of which is attached a blade having the form of that part of a circle subtended by 2 parallel chords; the blade passes through the diameter of the shaft so that the bottom of the blade is flush with the bottom of the shaft ; the shaft is placed so that its axis is within 2 mm of the axis of the vessel and the bottom of the blade is 25 ± 2 mm from the inner bottom of the vessel ; the upper part of the shaft is connected to a motor provided with a speed regulator; the stirrer rotates smoothly without significant wobble; 3. a water-bath that will maintain the dissolution medium at 37 ± 0.5 °C. Method. Place the prescribed volume of dissolution medium in the vessel, assemble the apparatus, warm the dissolution medium to 37 ± 0.5 °C and remove the thermometer. Place one unit of the preparation to be examined in the apparatus. Start the rotation of the apparatus immediately at the prescribed rate (± 4 %). Sampling and evaluation. Withdraw at the prescribed time, or at the prescribed intervals or continuously, the prescribed volume or volumes from a position midway between the surface of the dissolution medium and the top of the basket or blade and not less than 10 mm from the vessel wall. Except where continuous measurement is used with the paddle or basket method (the liquid removed being returned to the vessel) or where a single portion of liquid is removed, add a volume of dissolution medium equal to the volume of liquid removed or compensate by 31 calculation. Filter the liquid removed using an inert filter of appropriate pore size that does not cause significant adsorption of the active ingredient from the solution and does not contain substances extractable by the dissolution medium that would interfere with the prescribed analytical method. Proceed with analysis of the filtrate as prescribed. The quantity of the active ingredient dissolved in a specified time is expressed as a percentage of the content stated on the label. [17] 1.6. Model Drugs 1.6.1. Caffeine Coffeinum (Ph. Eur.) Figure 11. Formula of Caffeine. [17] Description. Caffeine contains not less than 98.5 % and not more than the equivalent of 101.5 % of 1,3,7-trimethyl-3,7-dihydro-1H-purine-2,6-dione, calculated with reference to the dried substance. A white, crystalline powder or silky, white crystals, sublimes readily, sparingly soluble in water, freely soluble in boiling water, slightly soluble in ethanol. It dissolves in concentrated solutions of alkali benzoates or salicylates. [17] Indications: drowsiness, fatigue, neonatal apnea. Mechanism. The most excepted explanation for caffeine‘s acute effects now is adenosine hypothesis. Adenosine is an inhibitory neurotransmitter. Caffeine and other methylxanthines occupy adenosine receptors and block the action. Effects. Primary action is stimulation of central nervous system activity. But there are actions outside the CNS: contraction of striated muscle, including the heart; relaxation of smooth muscle, especially the coronary arteries, uterus and bronchi; stimulation of gastric acid; diuretic effect; at higher doses, a stimulating effect on respiration; elevation of basal metabolism. Pharmacokinetics. Caffeine is rapidly absorbed from the GIT. The drug quickly reaches the brain because it can pass through the blood-brain barrier. The half-life of caffeine in the blood varies among people from 2.5 to 7.5 hours. Peak levels of caffeine occur 15 – 45 minutes after the drug is taken. Caffeine is equally distributed in total body water, so the concentration of 32 the drug is similar. It is metabolized primarily in the liver and is almost entirely excreted in the urine. [43] 1.6.2. Diltiazem Hydrochloride Diltiazemi hydrochloridum (Ph. Eur.) Figure 12. Formula of Diltiazem Hydrochloride. [17] Description. DH contains not less than 98.5 % and not more than the equivalent of 101.0 % of (2S,3S)-5-[2-(dimethylamino)ethyl]-2-(4-methoxyphenyl)-4-oxo-2,3,4,5-tetrahydro-1,5- benzothiazepin-3-yl acetate hydrochloride, calculated with reference to the dried substance. It is a white, crystalline powder, freely soluble in water, in methanol and in methylene chloride, slightly soluble in ethanol. It melts at about 213 °C with decomposition. [17] Indications: hypertension; prophylactic therapy for effort and vasospastic angina; supraventricular tachycardia. Mechanism. DH is a benzothiazepine calcium channel antagonist with proved antianginal and antihypertensive efficacy. Calcium channel-blocking agents produce a blockade of L-type (slow) calcium channels, which decreases contractile force and oxygen requirements. DH cause coronary vasodilatation and relief of spasm; it also dilate peripheral vasculature and decrease cardiac afterload. DH reduces the rate and contractility of the heart. Because it blocks calcium-dependent conduction in the atrioventricular node, DH can be used to treat atrioventricular nodal arrhythmias. Pharmacokinetics. DH is well-absorbed orally and undergoes hepatic oxidative metabolism. It is predominantly deacetylated into minimally active metabolite, which is then eliminated via the biliary tract. The half-life in plasma is approximately 3.0 – 4.5 h. [44] 33 1.7. Excipients 1.7.1. Microcrystalline Cellulose Cellulosum microcrystallinum (Ph. Eur.) Synonyms: Avicel PH; Celex; cellulose gel; Emcocel; Tabulose and etc. Description. MCC is purified, partially depolymerized cellulose that occurs as a white, odorless, tasteless, crystalline powder composed of porous particles. It is practically insoluble in water, in acetone, in ethanol, in toluene and in dilute acids. It is commercially available in different particle sizes, moisture, flow and other physical properties. The nominal mean size of Avicel PH-101 particle is 50 µm and moisture content is less than 5 %. Applications. MCC is used in pharmaceuticals, first as a binder (diluent) in tablet formulations where it is used in direct compression and wet granulation processes. MCC has also some lubricant properties. [17, 42] 1.7.2. Magnesium Stearate Magnesii stearas (Ph. Eur.) Synonyms: Magnesium octadecanoate; octadecanoic acid, magnesium salt; stearic acid, magnesium salt. Description. MgS is a mixture of magnesium salts of different fatty acids consisting mainly of stearic acid [(C17H35COO)2Mg; Mr 591.3] and palmitic acid [(C15H31COO)2 Mg; Mr 535.1] with minor proportions of other fatty acids. It contains not less than 4.0 % and not more than 5.0 % of Mg (Ar 24.30), calculated with reference to the dried substance. The fatty acid fraction contains not less than 40.0 % of stearic acid and the sum of stearic acid and palmitic acid is not less than 90.0 %. MgS is very fine, light white, precipitated or milled, impalpable powder of low density. It is practically insoluble in water and in ethanol. The powder is greasy to touch and readily adheres to skin. Applications. MgS is used as a lubricant in tablet manufacture at concentrations between 0.25 % and 5.0 %. Comments. MgS is hydrophobic and may retard the dissolution of a drug from a tablet; the lowest possible concentration is therefore used in manufacturing. Tablet dissolution rate and crushing strength decreases as the time of blending increases; and MgS may also increase tablet friability. Therefore the blending time with MgS should be controlled. [17, 42] 34 1.7.3. Colloidal Silicon Dioxide Silica colloidalis anhydrica (Ph. Eur.) Synonyms: Aerosil; fumed silica, Cb-O-Sil, colloidal silica and etc. Description. Colloidal Silicon Dioxide contains not less than 99.0 % and not more than the equivalent of 100.5 % of SiO2, determined on the ignited substance. CSD is a submicroscopic fumed silica with a particle size of about 15 nm. It is a light, loose, bluish-white colored, odorless, tasteless, nongritty amorphous powder. It is practically insoluble in water and in mineral acids except hydrofluoric acid. It dissolves in hot solutions of alkali hydroxides. Applications. Small particle size and large specific surface area give it desirable flow characteristics that are exploited to improve the flow properties of dry powders. [17, 42] 35 2. EXPERIMENTAL PART 2.1. Drugs and excipients Diltiazem hydrochloride, Zentiva, a.s., Czech Republic. Caffeine, Jilin Province Shulan Synthetic Pharmaceutical Co., Ltd, China. Avicel® PH 101 (Cellulosum microcrystalline), FMC Biopolymers, United States of America. Eudragit® NM 30 D, Evonik Röhm GmbH, Germany. Colloidal Silicon Dioxide, Degussa, Vicenza, Italy. Magnesium stearate, Peter Greven, Germany 2.2. Laboratory equipment Balance KERN 440-47, KERN & Sohn GmbH, Germany Analytical balance KERN 870-13, KERN & Sohn GmbH, Germany Optical microscope DN 25 Lambda, Intarcho-micro, Czech Republic CCD camera Alphaphot-2, Nikon, Japan Lamp Euromex Fiber Optic Light Source EK-1, Euromex Microscopes, Netherlands High shear mixer ROTOLAB machine, Zanchetta, Italy Drying oven Horo 048B, Dr. Ing. Hofman, Germany Equipment for sieve analysis Retsch AS 200 basic, RETSCH GmbH & Co., Germany Equipment to measure the flow ERWEKA SVM 102, ERWEKA GmbH, Germany Equipment for test of flowability MEDIPO, Czech Republic Mixer TURBULA T2C, Willy A. Bachofen, Switzerland Helium pycnometer Pycnomatic ATC, Porotec Vertrieb von Wissenschaflichen geräten GmbH, Germany Tablet press KORSCH EK 0, Korsch, Germany Tablet hardness tester C50 tablet Hardness & Compression tester, Engineering System (Notth) United Kingdom Friability tester ERWEKA TAR 10, ERWEKA GmbH, Germany Dissolution paddle apparatus SOTAX AT 7 Smart, SOTAX, Switzerland Spectrophotometer UV/VIS spectrophotometer, Perkin Elmer instruments, United States of America. 36 2.3. Preparation of granules 2.3.1. The measurement of particle size 0.01 g of diltiazem hydrochloride were spread on a glass slide. The diameter of 100 particles was measured using optical microscope connected with CCD camera. The same was done with caffeine and Avicel® PH 101. The obtained data were treated statistically by means of Microsoft Office Excel 2007 program. It was calculated the average value, the maximum and minimum values and standard deviation. 2.3.2. Preparation of granules It was weighted the necessary amount of model drugs, Avicel® PH 101 and 30 % aqueous dispersion of Eudragit® NM 30 D (see Table 7 and Table 8). Table 7. The composition of powder mixtures with diltiazem hydrochloride for granulates preparation Name of sample 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 Diltiazem hydrochloride (g) 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 Avicel® PH 101 (g) 100 100 100 100 100 100 100 100 100 100 50 50 50 50 50 50 25 25 25 25 25 Eudragit® NM 30 D aqueous dispersion (g) 10.5 22.2 35.3 50 66.6 85.7 107.7 107.9 130.1 152.3 7.9 16.7 26.5 37.5 50 64.3 6.6 13.9 22.1 31.25 41.7 37 Table 8. The composition of powder mixtures with caffeine for granulates preparation Name of sample 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 Caffeine (g) 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 Avicel® PH 101 (g) 100 100 100 100 100 100 100 50 50 50 50 50 50 50 25 25 25 25 25 25 25 25 25 25 Eudragit® NM 30 D aqueous dispersion (g) 10.5 22.2 35.3 50 66.6 85.7 107.9 7.9 16.7 26.5 37.5 50 64.3 81 6.6 13.9 22.1 31.25 41.7 53.6 55.6 67.5 81.4 95.3 Granulates were prepared in high-shear mixer ROTOLAB (see Figure 13). The highshear mixer been set like this: impeller pause time – 0 sec, impeller working time – 300 s, cycle time – 300 s, impeller speed – 1200 Rpm. Diltiazem hydrochloride and Avicel PH 101 was mixed for first 30 sec without polymer, then other 30 seconds the binder liquid was added manually and the mixture was blended for 240 s. The mixture was passed through 1.25 mm mesh sieve and granules were dried for 24 h at 40° C in a drying oven. After drying granules were passed trough 1.25 mm sieve again. The granulation of batches with Eudragit® NM 30 D (aqueous dispersion) amount till 30 % from the mass of granulation (in case of 1-7, 11-21, 22-27, 29-34 samples) or 25 % (in case of 36-40 samples) was made by one step. The granulation with higher amount of Eudragit® NM 30 D (8-10, 28, 35, 41-45 samples) was made by several steps. First step it was adding 25 % (or 30 %) of Eudragit® NM 30 D, passing through the sieve, drying. The second step it was adding next 10 % portion of Eudragit® NM 30 D to the granules, passing through the sieve, drying. If it was necessary third and fourth granulation step (next 10 % portion of Eudragit® NM 30 D) was made till desired concentration of Eudragit® NM 30 D. 38 The granulation by steps helps to avoid to over wet of granules. The obtained granules were tested to determine their suitability for tablet compression process. Figure 13. High-shear mixer ROTOLAB. 2.4. Evaluation of granules quality parameters 2.4.1. Determination of granules flowability 100 g of sample was introduced without compacting into a dry funnel, whose bottom opening blocked by suitable means. Nozzle Nr. 3 (25 mm) was used. The bottom opening was unblocked and the time needed for the entire sample to flow was measured. Test of flowabilty was repeated three times. Results were expressed in seconds and tenths of seconds. The samples, which were not flowing at all, were not tested by other tests. 2.4.2. Determination of granules flow (Compressibility index and Hausner ratio) This test is intended to determine under defined conditions the apparent volumes, before and after settling, the ability to settle and the apparent densities of granules. [17] Samples of granules which did not had the necessary flowability were not tested. Into the dry 100 ml cylinder it was introduced without compacting approximately 35 g of sample. The unsettled apparent volume (V0) was read to the nearest milliliter. 1250 taps was 39 made by ERWEKA apparatus and the final tapped volume was read to the nearest milliliter. This test was repeated three times to avoid inaccuracy. The results were calculated and expressed as the average of bulk density, tapped density, compressibility index and Hausner ratio and standard deviation according to 1, 2, 3, 4 formula (see Page 28). 2.4.3. Determination of density using helium-pycnometer It was determined the real density of granules samples which had necessary flowability. The helium-pycnometer POROTEC was turned on. A dry metal vessel was weighted accurately using the analytical balance. It was filled approximately 10.0 g of sample and it was weighted again accurately. The vessel with sample was placed into a chamber. The helium gauge was opened. The weight of empty and loaded vessel were entered to the helium-pycnometer. When the temperature was 20° C, the measurement was started. Results were expressed as density (g/cm3) and it was calculated standard deviation. 2.4.4. Determination of granules size using sieve analysis Sieve analysis was used to determine the percentage of size distribution of granules particles. Batches of granules which did not had necessary flowability were not tested. 100 g of sample were sieved for 10 min and 60 amplitude on a set of sieves with meshes sizes from 1.25, 1.00, 0.8, 0.5, 0.25, 0.125 and 0.08 mm using a vibrating sieving equipment. The results were expressed as percents of granules portions. 2.5. Preparation of matrix tablets 2.5.1. Preparation of granules for compressing After tests to determine the suitability of granules for compressing were selected samples which have necessary properties for preparing tablets. After sieve test the granules were not homogenous, so they were mixed for 2 min in TURBULA. Then it was added 0.5 % MgS and 0.5 % CSD from the weight of granules to each sample (see Table 9 and 10). CSD was passed through 1.25 mm sieve, because the powder was not homogenous, it had some agglomerates. Granules with excipients were blended for 5 min in TURBULA. 40 2.5.2. Compression of matrix tablets The samples were pressed into a matrix tablets using eccentric tablet press KORSCH, it was used 10 mm punches. Tablets were made like that, that each tablet would contain 100 mg model drug. The composition of matrix tablets is shown in Table 12 and Table 13. Table 9. The composition of matrix tablets with diltiazem hydrochloride. Sample Diltiazem hydrochloride (mg) Avicel® PH 101 (mg) Eudragit® NM (mg) Magnesium Stearate (mg) Colloidal Silicon Dioxide 1D 2D 3D 4D 5D 6D 7D 8D 9D 10D 11D 12D 13D 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 50 50 50 50 25 25 25 15 19,98 25,71 32,37 39,03 45,69 7,95 11,25 15 19,29 6,63 9,375 12,51 1,085 1,11 1,14 1,17 1,21 1,24 0,8 0,8 0,83 0,86 0,66 0,68 0,7 1,085 1,11 1,14 1,17 1,21 1,24 0,8 0,8 0,83 0,86 0,66 0,68 0,7 (mg) Table 10. The composition of matrix tablets with caffeine Sample 1C 2C 3C 4C 5C 6C 7C 8C 9C 10C 11C 12C 13C 14C 15C 16C 17C Caffeine (mg) 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 Avicel® PH 101 (mg) 100 100 100 100 50 50 50 50 50 25 25 25 25 25 25 25 25 Eudragit® NM (mg) 15 19,98 25,71 32,37 7,95 11,25 15 19,29 24,3 6,63 9,375 12,51 16,08 16,68 20,25 24,42 28,59 Magnesium Stearate (mg) 1,085 1,11 1,14 1,17 0,8 0,8 0,83 0,86 0,88 0,66 0,68 0,7 0,71 0,72 0,73 0,75 0,78 Colloidal Silicon Dioxide (mg) 1,085 1,11 1,14 1,17 0,8 0,8 0,83 0,86 0,88 0,66 0,68 0,7 0,71 0,72 0,73 0,75 0,78 41 2.6. Evaluation of quality parameters of matrix tablets (Ph. Eur.) 2.6.1. Uniformity of tablet mass It was weighted individually 20 tablets taken at random from each tablet set by the analytical balance. It was determined the average mass value expressed by milligrams, the deviation of ±7.5 % from average mass and standard deviation. 2.6.2. Uniformity of tablet content The uniformity of tablet content was determined by spectrophotometric method measuring the absorbance of sample. UV/UVIS spectrophotometer Perkin Elmer instruments was used for the measurement. The wavelength to measure the absorbance was 237 nm for samples with diltiazem hydrochloride and 275 nm for samples with caffeine. It was taken 10 tablets by random from each sample. Each tablet was crushed and approx. 50 mg of the powder was quantitatively transferred into 100 ml flask and it was filled with distilled water till the line. After 24 hours 1 ml of solution was taken and transferred into other 100 ml flask and filled with distilled water till the line again and the solution was mixed. The concentration of solution was 0.0001 g/l. The sample was filtrated and then the absorbance was measured. 100 mg model drug (diltiazem hydrochloride or caffeine) was taken to prepare the standard solution. It was dissolved in 100 ml distilled water. After 24 hours 1 ml of solution was transferred into 100 ml flask and it was filled with distilled water till 100 ml. The concentration of standard was 0.0001 g/l. The standard solution was filtrated before measurement. The content of model drug in tablets was calculated according to the formula 5: X Asa mtbl mst d sa Ast msa d st (5) where: X – drug amount in tablet (g), Asa – absorbance of studied solution, Ast – absorbance of standard solution, mtbl – mass of tablet (g) dsa – dilution of tested sample dst – dilution of standard sample msa – mass of tested sample (g) 42 mst – mass of tested sample (g) The average content of model drug from in each sample was calculated of 10 measurement values and the deviation of ±15 % from the each average content. The results were expressed by the average value and standard deviation. 2.6.3. Friability of tablets The sample for test was taken by random from each tablet set. The tablets were accurately degusted before weighting. The sample weight was near as possible to 6.5 g. The sample was placed into a friability apparatus ERWEKA drum. The drum was rotated 100 times for 4 minutes (25 rotations per minute). The tablets were removed from the drum; sample was cleaned from the dust as before and accurately weighted by the analytical balance. Test for each sample was made once. The sample mass was expressed in milligrams. Abrasion of tablets was expressed as the percentage loss from the initial mass of sample. 2.6.4. Resistance to crushing of tablets (Hardness of tablets) It was taken 10 tablets by random from each sample. Tablets hardness was measured using C50 Tablet & Compression Tester. Tablet was placed in the radial direction between jaws of apparatus for automatic measurement. After measurement all fragments of tablet is removed by brush. Results were expressed as the average, minimum and maximum values in Newton of the used force. 2.6.5. Determination of released drug from matrix tablet The dissolution test was proceeded in paddle apparatus SOTAX AT7 Smart, which is the part of automatic dissolution line. The dissolution medium was selected phosphate buffer solution pH 6.8 R1 (Ph. Eur.). It consists from 51.0 ml of a 27.2 g/l solution of potassium dihydrogen phosphate and 49 ml of a 71.6 solution of disodium hydrogen phosphate. It was measured 1000 ml of dissolution medium in each cylindrical vessel, which were placed into a water-bath that is maintaining the temperature of dissolution medium at 37 ± 0.5 °C. Individual tablets automatically fell to the bottom of vessel. The speed of stirrer was 100 rounds per minute. The sample of dissolution medium with released model drug was taken automatically at the time intervals 30, 60, 120, 180, 240, 300, 360, 420, 480, 540, 600, 660 and 720 minutes. The amount of released model drug was measured by UV/UVIS spectrophotometer Perkin Elmer instruments. The amount of released drug in the prescribed time interval was expressed by percents. 43 The dissolution test with continual pH change was performed at 37±0.5 ◦C in 900 ml of buffer with pH 1.2 (artificial gastric juice – AGJ) for 2 hours. Sodium triphosphate was used as the pH increasing agent. After 2 hours interval, the pH value was changed to 6.8 by adding of 18.7 g of sodium triphosphate for following 10 hours. The stirring rate was of 100 rpm. 44 3. RESULTS AND DISCUSSION 3.1. Preparation of granules Two drugs were selected as the model drugs: freely soluble in water diltiazem hydrochloride and sparingly soluble in water caffeine. Microcrystalline cellulose was used as the insoluble diluent. DH, C and MCC particle size were evaluated by image analysis (optical microscope connected to CCD camera). The particle size distribution of model drugs and MCC are shown in Figures 15, 17, 19 and their stereomicroscopic photographs in Figures 14, 16, 18. The main fraction (66.5 %) of DH particles size was between 41 and 80 µm, 18 % of particles were smaller than 40 µm, 15 % of particles were between 81 and 120 µm and less than 1% of particles were bigger than 121 µm. Differences of DH particle size and its distribution were more significant than C or MCC particles. The main fraction (53 %) of C particles size was between 41 and 80 µm, 15.5 % of particles were smaller than 40 µm, 23 % of particles are 81-120 µm and 10.5 % of particles were bigger than 121 µm. The size distribution of C and MCC particles was more uniform in all intervals in comparison with DH particles size distribution. The size distribution of C particles differed from MCC one in amount particles bigger than 121 µm. The uniformity of particles size distribution (between DH and MCC or C and MCC) ensured tantamount mixing during the technological processes. It was prepared three DH sets which differed by the ratio of model drug and MCC, the ratio between DH and MCC was 1:1, 2:1 and 4:1. Sets of C were made according to the same ratio. Wet granulation was performed in high shear mixer by adding of different amount of Eudragit® NM 30 D (for composition see Tables 7 and 8). Less than 10 % amount Eudragit® of NM 30 D aqueous dispersion did not ensure the formation of DH and MCC or C and MCC granules, because the amount of binder was too low. The granulation with more than 30 % amount of Eudragit® NM 30 D was made by several steps to avoid forming of wet mass which can not be meshed through the sieve. By first step of study, maximal amount of polymer was 30 % (aqueous dispersion) in granules of all sets DH and C. Granules were evaluated, then matrix tablets were pressed, evaluated by test of Ph. Eur., but drug release was too fast. Trend between the amount of drug and MCC and polymer was observed and one set with DH and one set with C have been extended with adding higher amount of Eudragit® NM 30 D. Total it was made 21 DH and 24 C different granulates. 45 3.2. Results of granules evaluation The flowability time of DH or C granules is relatively small, so it provides good filling of dies during tablet manufacturing. The flowability of DH different granules is quite similar. The flowability of samples, which ratio between DH and MCC was 1:1 (1D – 6D, see Figure 20), differed less than 1 second. The same time differences were found between samples with ratio DH and MCC 2:1 (7D – 10D see Figure 21) or 4:1 (11D – 13D see Figure 22). Increasing amount of Eudragit® NM in granules did not provide the significant change of flowability time but low decrease of flowability time was observed as the trend. The amount of MCC was not significant parameter for flowability. Almost the same difference of flowability time (less than 1.5 second) of C granules batches with different amount of MCC, so the amount of MCC did not influence time. Higher amount of Eudragit® NM in C granules did not provide significant changes too. The flowability time was observed to be slightly lower with increasing concentration of Eudragit® NM. The flow character of DH granules sets varied from passable to excellent. In all DH sets of samples flow character reached “excellent” rating, so the ratio between DH and MCC is not most important parameter (see Table 13). The main influence had the amount of Eudragit® NM. Samples having low concentration of Eudragit® NM had higher compressibility index than samples with high concentration. Probably this is due to the bigger size of prepared granules and physicochemical properties of DH. It is well known that the higher proportion of a binder used for granulation process leads to the enlargement of granulate particle diameter [46]. Large granules possess better flow properties and it ensures good die filling during tablet manufacture. The flow character of C granules sets varied not much (see Table 14). In first set of C granules (ratio between C and MCC is 1:1) flow character is passable, in second set (ratio of C and MCC is 2:1) flow character changed from passable till fair and in the third set (ratio of C and MCC is 4:1) flow character changes from passable till fair. Probably compressibility index was influenced by the amount of Eudragit® NM and physicochemical properties of C. Higher amount of polymer improve flow character of C granules [34]. Different physicochemical properties of DH and C lead to a different Hausner ratio of these drugs granules, when the amount of MCC and Eudragit® NM is the same in granules. DH granules possess better flow character than C granules. This fact could be caused by very poor flow properties of starting material – caffeine. A flow rate is lower than 10 mg/s. [46] For this reason, granules of C possess worse flow properties than DH granules. 46 Pycnometric density of DH granules decreased in all sets of samples. Amount of polymer influence and MCC influence this. Density decrease was probably due to higher amount of Eudragit® NM which led to bigger and thicker granules formation with higher air amount. Granules having lower quantity of MCC possessed lower density than samples with higher amount of MCC. The same trend was observed between the amount of polymer and MCC and density of C. Pycnometric density of all C granules was higher than DH ones. For results of pycnometric density see Table 15 for DH granules and Table 16 for C granules. Size of DH granules was influenced by amount of Eudragit® NM. Samples with small concentration of polymer had bigger amount of small size granules (from 0,125 mm and smaller) and samples with higher amount of Eudragit® NM had little amount of small size granules. Higher amount of polymer led to formation of bigger granules. The same trend was observed in the case of C granules (see Table 17 and 18). Results of granules evaluation could be considered as similar between DH and C. 3.3. Results of tablets evaluation The weights of all DH and C matrix tablets were in accordance with the European Pharmacopoeia limits (see Table 19 and 20). This is due to good flowing properties of granules. The model drugs content in matrix tablets were between limits of European Pharmacopoeia (see Table 19 and 20) with maximal SD 5.87. All matrix formulations were compliant with official friability limit. The friability was very small, it was less than 0,1 % for DH matrix tablets (see Table 21) and less than 0,2 % for C tablets (see Table 22). No significant differences between samples friability were observed. Hardness of DH matrix tablets were from 107,27 N (11D) to 161,13 N (6D). Lower amount of polymer in tablets led to similar hardness of tablets in a set and small deviation from average value. Higher amount of Eudragit® NM in tablet composition resulted in improving of tablet mechanical properties (see Figure 26). Tablet with high percentage of polymer was resistance to crushing, first it deformed and when the crushing force became critical tablet cracks. For this reason the deviation from average value was high among samples of set. Hardness of C matrix tablets were from 107,71 N (1C) to 134,46 (15C) - see Figure 27. Higher amount of Eudragit® NM led to higher standard deviation values of hardness average mean between samples of set. C matrix tablets are not so plastic as DH matrix tablets and this is due to different physicochemical properties of drug. 47 Compression force of DH tablets (see Figure 28) was various depending from drug and MCC ratio. Sets with lower amount of MCC needed higher compression force to produce tablets with similar hardness in comparison with samples with higher amount of MCC, for example the compression force of 1D is 10,53 kN and average of tablet hardness is 108,11 N, compression force of 11D is 31,59 kN and average of tablet hardness is 107,27 N. The amounts of polymer in tablets and their hardnesses were similar, but compression forces were different. So the ratio between drug and MCC is crucial parameter for compression force. There is the same trend observed with compression force of C tablets (see Figure 29). Higher compression force was necessary to be used to achieve similar hardness of tablets with low amount of MCC comparing with samples having higher amount of MCC. This is due to good compressibility properties of MCC, which reduces the total compression force. In general, compression force of C tablets is lower than compression of DH tablets. Results of tablet evaluation are similar between DH and C matrix tablets. The dissolution profile of DH matrix tablets was various depending from amount of MCC and Eudragit® NM (see Figure 30, 31, 32, 33). The dissolution rate of samples with low amount of polymer is fast, due to insufficient layer thickness of matrix. Increasing amount of Eudragit® NM in matrix tablet showed more satisfactory retardation of drug release. Ratio between DH and MCC is also important parameter for matrix formulation. When ratio was 1:1 dissolution profile was more gradual. Burst effect defined as the faster drug release from tablet surface in the beginning of dissolution [39], was high in all samples (about 30 %), so it would be recommended to coat these tablets to make dissolution profile more gradual. The dissolution profile of C matrix tablets was unexpected, because almost all samples of tablets disintegrated during first hours and did not show almost any retardation of drug release (see Figure 34, 35, 36, 37, 39). This is probably due to physicochemical properties of drug components. C is slightly soluble in water, MCC is almost insoluble in water and Eudragit® NM is insoluble too, so it can cause disintegration of tablets. One sample (14C) showed retardation of C release. The same sample of 14C tablet was prepared twice using the same components to avoid mistakes. Both samples of 14C demonstrated similar dissolution profile. So it is optimal amount of polymer and MCC for C matrix tablet. In future study, it could be recommended to use other diluent not MCC. Samples which showed optimal release profile (4D, 5D and 14C) were tested with dissolution test with continual pH change for 2 hours at pH 1.2 and later at pH 6.8 (see Figure 39, 40). The release profile was to fast at pH 1.2, so tablets are recommended to coat acidoresistant coating (Eudragit® L 30 D). 48 CONCLUSIONS 1. Wet granulation method using high-shear mixer and Eudragit® NM 30 D is suitable to produce DH and C granules. 2. It was found, that minimal amount of polymer has to be 5 %, which ensures forming of granules possessing suitable properties for tablets manufacturing. 3. Eudragit® NM is suitable for manufacturing of DH and C matrix tablets, which are in accordance with limits of tablet test of Ph. Eur. 4. It was found optimal amount of MCC and polymer in tablets, which leads to gradual release of drug for 12 hours at pH 6.8. In case of DH, the ratio between drug and MCC has to be 1:1 and contain 13 – 16 % of polymer in tablet. In case of C, the ratio has to be 4:1 and contain 12 % of Eudragit® NM in tablet. 49 REFERENCES 1. McGinity JW, Felton LA. Aqueous polymeric coatings for pharmaceutical dosage forms. 3rd ed. New York: Informa Healthcare USA; 2008. 109, 238, 258, 267 2. Moustafine RI, Kabanova TV, Kemenova VA, Mooter GV. Characteristics of interpolyelectrolyte complexes of Eudragit E100 with Eudragit L100. J. of Control. Release 103 (2005) 191-198. 3. Eudragit® product brochure. Available at: http://www.pharma-polymers.com/NR/rdonlyres/3D19A053-7628-41B1-BC52123763B75566/0/090821FinalEUDRAGIT_online.pdf 4. Specifications and test methods for Eudragit® L 30 D-55. Available at: http://www.pharmapolymers.com/pharmapolymers/MCMSbase/Pages/ProvideResource.a spx?respath=/NR/rdonlyres/63F690A0E7834936B2D0676A82FD102B/0/INFO705_E_L 30D55.pdf 5. Specifications and test methods for Eudragit® 100-55. Available at: http://www.pharmapolymers.com/pharmapolymers/MCMSbase/Pages/ProvideResource. aspx?respath=/NR/rdonlyres/A6B556A563D141D4BF73CD75DD83955C/0/INFO704_ E_L10055.pdf 6. Specifications and test methods for Eudragit® L 100 and Eudragit® S 100. Available at: http://www.pharmapolymers.com/pharmapolymers/MCMSbase/Pages/ProvideResource. aspx?respath=/NR/rdonlyres/E90B3F4073254E2283D3285B42CC601B/0/INFO703_E_ L100_S100.pdf 7. Specifications and test methods for Eudragit® L 12,5 and Eudragit® S 12,5. Available at: http://www.pharmapolymers.com/pharmapolymers/MCMSbase/Pages/ProvideResource.a spx?respath=/NR/rdonlyres/A2C6A670873F45BAA5A85C2109A731DB/0/INFO708_E _RL125_RS125.pdf 8. Specifications and test methods for Eudragit® FS 30 D. Available at: http://www.pharmapolymers.com/pharmapolymers/MCMSbase/Pages/ProvideResource. aspx?respath=/NR/rdonlyres/EA96FF4EBB7441488E29631B73E3B6F0/0/Specification FS30D_xeri21B86.pdf 9. Specifications and test methods for Eudragit® RL 100 and Eudragit® RL PO, Eudragit® RS 100 and Eudragit® RS PO. Available at: http://www.pharmapolymers.com/pharmapolymers/MCMSbase/Pages/ProvideResource.aspx?respath=/Rdo 50 nlyres/6B60AAA0CD0D49D69FCFB4F6CC2F736D/0/INFO707_E_RL100_RLPO_RS 100_RSPO.pdf 10. Specifications and test methods for Eudragit® RL 30 D and Eudragit® RS 30 D. Available at: http://www.pharmapolymers.com/pharmapolymers/MCMSbase/Pages/ProvideResource. aspx?respath=/NR/rdonlyres/AAF66925A5854D20836BD9D99F1EDC9A/0/INFO709_ E_RL30D_RS30D.pdf 11. Specifications and test methods for Eudragit® RL 12,5 and Eudragit® RS 12,5. Available at: http://www.pharmapolymers.com/pharmapolymers/MCMSbase/Pages/ProvideResource. aspx?respath=/NR/rdonlyres/A2C6A670873F45BAA5A85C2109A731DB/0/INFO708_ E_RL125_RS125.pdf 12. Specifications and test methods for Eudragit® NE 30 D. Available at: http://www.pharmapolymers.com/pharmapolymers/MCMSbase/Pages/ProvideResource. aspx?respath=/NR/rdonlyres/343EEF663DDC4C29BDFD31600B26A183/0/INFO706_ E_NE30D.pdf 13. Specifications and test methods for Eudragit® NE 40 D. Available at: http://www.pharmapolymers.com/pharmapolymers/MCMSbase/Pages/ProvideResource. aspx?respath=/NR/rdonlyres/94E67D3937304BC58F642692B0939495/0/INFO71_E_N E40D.pdf 14. Specifications and test methods for Eudragit® NM 30 D. Available at: http://www.pharmapolymers.com/pharmapolymers/MCMSbase/Pages/ProvideResource. aspx?respath=/NR/rdonlyres/348C25416BFD4A17AA66A389111FCEA4/0/INFO714_ E_NM30D.pdf 15. Brögmann B, Beckert TE. Enteric targeting through enteric coating. In: Schreier H. Drug targeting technology. New York: Marcel Dekker; 2001. 1,6 16. Colo DG, Falchi S, Zambito Z. In vitro evaluation of a system for pH-controlled peroral delivery of metformin. J. of Control. Release 80 (2002) 119-128. 17. European Pharmacopoiea 5.0. Volumes I and II. Strasbourg: Council of Europe; 2005. 228-230, 233-235, 239, 241-243, 626-628, 1145, 1228-1232, 1443-1444, 1961-1962, 2005-2007. 18. Kendall RA, Alhnan MA, Nilkumhang S, Murdan S, Basit AW. Fabrication and in vivo evaluation of higly pH-responsive acrylic microparticles for targeted gastrointestinal delivery. European Journal of Pharmaceutical Sciences 37 (2009) 284-290. 51 19. Fan TY, Wei SL, Yan WW, Chen DB, Li J. An investigation of pulsatile release tablets with ethylcellulose and Eudragit L as film coating materials and cross-linked polyvinylpyrrolidone in the core tablets. J. of Control. Release 77 (2001) 245; 245-251 20. Chan WA, Boswell CD, Zhang Z. Comparison of the release profiles of a water soluble drug carried by Eudragit-coated capsules in different in-vitro dissolution liquids. Powder technology 119 (2001) 26-32 21. Ibekwe VC, Fadda HM, Parsons GE, Basit AW. A comparative in vitro assessment of the drug release performance of pH-responsive polymers for ileo-colonic delivery. International Journal of Pharmaceutics 308 (2006) 52-60 22. Miller DA, McGinity JW. Aqueous polymeric film coating. In: Augsburger LL, Hoag SW. Pharmaceutical dosage forms: tablets. 3rd ed. Volume 1: Unit operations and mechanical properties. New York: Informa Healthcare USA; 2008. 414-421, 430 23. Akhgari A, Farahmand F, Afrasiabi GH, Sadeghi F, Vandamme TF. Permeability and swelling studies on free films containing inulin in combination with different polymethacrylates aimed for colonic drug delivery. European Journal of Pharmaceutical Sciences 28 (2006) 307-314. 24. El-Malah Y, Nazzal S. Novel use of Eudragit® NE 30 D/Eudragit® L 30D-55 blends as functional coating materials in time-delayed drug release applications. International Journal of Pharmaceutics 357 (2008) 219-227. 25. Colombo P, Santi P et al. Swellable and Rigid Matrices: Controlled Release Matrices with Cellulose Ethers. In: Augsburger LL, Hoag SW. Pharmaceutical dosage forms: tablets. Volume 2: Rational Design and Formulation. 3rd ed. New York: Informa Healthcare USA; 2008. 432-437. 26. Dosage forms with a drug attached to a polymer dispersed in a non-erodible polymer matrix. In: Vergnaud JM. Controlled drug release of oral dosage forms. Chichester; Ellis Horwood Limited; 1993. 394 27. Ceballos A, Cirri M, Maestrelli F, Corti G, Mura P. Influence of formulation and process variables on in vitro release of theophylline from directly-compressed Eudragit matrix tablets. Il Farmaco 60 (2005) 913-918. 28. El-Shanaway S. Sustained release of nitrofurantoin from inert wax matrices. J. of Control. Release 26 (1993) 11-19. 29. Arevalo MF, Villafuerte MAH, Dorado JMG, Alvarez AMR. Effects of different fillers and wetting liquids on the dissolution of carteolol hydrochloride controlled release inert matrix tablets. International Journal of Pharmaceutics 95 (1993) 117-125 52 30. Katzhendler I, Hoffman A, Goldberger A, Friedman M. Modeling of Drug release from Erodible Tablets. Journal of Pharmaceutical Sciences 86 (1997) 110-115 31. Karasulu HY, Ertan G, Köse T. Modeling of theophylline release from different geometrical erodible tablets. European Journal of Pharmaceutics and Biopharmaceutics 49 (2000) 177-182 32. Swarbrick J. Encyclopedia of pharmaceutical technology. Volume 6. 3ed. New York; Informa Healthcare USA; 2007. 3653 - 3654, 3659 33. Swarbrick J, Boylan JC. Encyclopedia of pharmaceutical technology. Volume 3. 2ed. New York; Marcel Dekker; 2002. 2267, 2701-2706, 2717 34. Pereira de Souza T, Martínez-Pacheco R, Gómez-Amoza JL, Petrovick PR. Eudragit E as Excipient for Production of Granules and Tablets From Phyllanthus niruri L SprayDried Extract. AAPSPharmSciTech. 8 (2) (2007) E1-E7. 35. Eudragit® 3-Day Workshop. Hot Melt Extrusion & Modified Release Applications. Available at: http://www.pharmapolymers.com/pharmapolymers/MCMSbase/Pages/ProvideResource. aspx?respath=/NR/rdonlyres/9725600847D34DDAB4067ECAC96EDE70/0/09Pharma4 pgs8x11forEmailFallWorkshops.pdf 36. Gibson M. Pharmaceutical Preformulation and Formulation. A Practical Guide from Candidate Drug Selection to Commercial Dosage form. Boca Raton; CRC Press; 2004. 182-183 37. Gupta VK, Beckert TE, Price JC. A novel pH- and time-based multi-unit potential colonic drug delivery system. I. Development. International Journal of Pharmaceutics 213 (2001) 83-91. 38. Leopold CS. Coated dosage forms for colon-specific drug delivery. Pharmaceutical Science & Technology Today. Volume 2. 5 (1999) 197-204. 39. Arno EA, Anand P, Bhaskar K, Ramachandran S, Saravanan M, Vinod R. Eudragit NE30D Based Metformin/Gliclazide Extended Release Tablets: Formulation, Characterisation and in Vitro Release Studies. Chemical & Pharmaceutical Bulletin. Volume 50. 11 (2002) 1495-1498. 40. Yang WC. Handbook of Fluidization and Fluid-Particle Systems. New York: Marcel Dekker; 2003. 6-8. 41. Niazi SK. Handbook of Pharmaceutical Manufacturing Formulations: Uncompressed Solid Products. Volume 2. Boca Raton: CRC Press; 2004. 41-43 53 42. Rowe RC, Sheskey PJ, Owen SC. Handbook of Pharmaceutical Excipients. 5th ed. London: Pharmaceutical Press; 2006. 132-135, 188-191, 430-433. 43. Maisto SA, Galizio M, Connors GJ. Drug Use and Abuse. 5th ed. Belmont: Thompson Wadsworth; 2008. 168-184. 44. Trevor AJ, Katzung BG, Masters S. Katzung & Trevor’s Pharmacology. Examination and Board Review. 8th ed. Columbus: The McGraw-Hill Companies; 2008. 97-128. 45. Tvardovskiy AV. Sorbent Deformation. Oxford: Elsevier; 2007. 111-118. 46. Seppälä K, Heinämäki J, Hatara J, Seppälä L, Yliruusi J. Development of a New Methoc to Get a Reliable Powder Flow Chracterictics Using Only 1 to 2 g of Powder. AAPSPharmSciTech. 11 (1) (2010). 402-408. 54 ADDITION Nr. 1 Figure 14. Stereomicroscopic photographs of diltiazem hydrochloride particles. Figure 15. The particle size distribution of diltiazem hydrochloride. 55 Figure 16. Stereomicroscopic photographs of caffeine particles. Figure 17. The particle size distribution of caffeine. 56 Figure 18. Stereomicroscopic photographs of Avicel® PH 101. Figure 19. The particle size distribution of Avicel® PH 101 particles. 57 ADDITION Nr. 2 Figure 20. Flowability of granule’s mixture consisting of DH and MCC in ratio 1:1 and different amounts of NM 30D. Figure 21. Flowability of granule’s mixture consisting of DH and MCC in ratio 2:1 and different amounts of NM 30 3 2,5 Time (s) 2 1,5 1 0,5 0 7D 8D 9D 10D Sample 58 Figure 22. Flowability of granule’s mixture consisting of DH and MCC in ratio 4:1 and different amounts of NM 30D. 3 2,5 Time (s) 2 1,5 1 0,5 0 11D 12D 13D Sample Table 11. Standard deviation of flowability test accomplished with mixture of granules consisting DH, MCC and NM 30 D. Sample SD (s) 0,04 1D 0,04 2D 0,01 3D 0,01 4D 0,02 5D 0,01 6D 0,06 7D 0,03 8D 0,02 9D 0,02 10D 0,02 11D 0,02 12D 0,01 13D 59 Figure 23. Flowability of granule’s mixture consisting of C and MCC in ratio 1:1 and different amounts of NM 30D. 3,5 3 2,5 Time (s) 2 1,5 1 0,5 0 1C 2C 3C 4C Sample Figure 24. Flowability of granule’s mixture consisting C and MCC in ratio 2:1 and different amounts of NM 30D. 3,5 3 Time (s) 2,5 2 1,5 1 0,5 0 5C 6C 7C 8C 9C Sample 60 Figure 25. Flowability of granule’s mixture consisting C and MCC in ratio 4:1 and different amounts of NM 30D. 3,5 3 Time (s) 2,5 2 1,5 1 0,5 0 10C 11C 12C 13C 14C 15C 16C 17C Sample Table 12. Standard deviation of flowability test accomplished with mixture of granules consisting C, MCC and NM 30 D. Sample 1C 2C 3C 4C 5C 6C 7C 8C 9C 10C 11C 12C 13C 14C 15C 16C 17C SD (s) 0,08 0,02 0,02 0,02 0,03 0,01 0,03 0,03 0,02 0,03 0,02 0,02 0,02 0,01 0,01 0,01 0,02 61 Table 13. Flow test results: Hausner ratio and compressibility index calculations of prepared granules with diltiazem hydrochloride. Sample m (g) V0 (ml) Vf (ml) ρbulk (g/ml) ρtapped (g/ml) 1D 34,9 93 75,3 0,375 0,463 1,23 18,99 SD 0 1 0,6 0,004 0,004 0,01 0,46 2D 34,9 93 80 0,376 0,437 1,16 13,97 Hausner Compressibility ratio index (%) SD 0,1 1 1 0,004 0,006 0,02 1,72 3D 35 92 80,7 0,38 0,434 1,14 12,32 SD 0 1 1,5 0,004 0,008 0,01 0,75 4D 35 82 72,3 0,427 0,484 1,13 11,78 SD 0,1 1 0,6 0,004 0,004 0,02 1,28 5D 35,3 81,3 70,7 0,435 0,5 1,15 13,11 SD 0,1 0,6 0,6 0,004 0,004 0,01 0,67 6D 35,1 79 71,3 0,444 0,492 1,11 9,7 SD 0,2 1 1,5 0,005 0,008 0,02 1,89 7D 34,3 77,3 59 0,444 0,581 1,31 23,68 SD 0,7 2,3 1 0,008 0,002 0,02 1,32 8D 37,8 95 80,3 0,398 0,471 1,18 15,43 SD 0,2 1 0,6 0,003 0,004 0,02 1,07 9D 34,1 90,3 80,7 0,378 0,423 1,12 10,7 SD 0,1 0,6 0,6 0,002 0,003 0,01 0,61 10D 33,8 92 85,7 0,368 0,395 1,07 6,862 SD 0,3 2 1,5 0,009 0,01 0,02 2,19 11D 34,1 81,7 68,3 0,417 0,499 1,2 16,3 SD 0,4 2,1 0,6 0,006 0,003 0,02 1,45 12D 34,9 88,3 79,3 0,395 0,439 1,11 10,19 SD 0,3 0,6 0,6 0,003 0,003 0 0,07 13D 34,4 88,3 83,3 0,39 0,413 1,06 5,66 SD 0,3 2,1 2,1 0,005 0,006 0 0,13 Flow character Fair Good Good Good Good Excellent Passable Fair Good Excellent Fair Good Excellent 62 Table 14. Flow test results: Hausner ratio and compressibility index calculations of prepared granules with caffeine. Sample m (g) V0 (ml) Vf (ml) ρbulk ρtapped Hausner Compressibility (g/ml) (g/ml) ratio index (%) 1C 36,4 80,7 61,3 0,451 0,593 1,32 23,97 SD 0,2 0,6 0,6 0,002 0,004 0,01 0,65 2C 36,6 84,3 66,3 0,434 0,552 1,27 21,34 SD 0,2 0,6 0,6 0,001 0,003 0 0,15 3C 36,9 89,3 70,3 0,413 0,525 1,27 21,26 SD 0,2 1,2 0,6 0,004 0,002 0,01 0,89 4C 35,4 79,7 63,3 0,445 0,559 1,26 20,5 SD 0,2 1,2 0,6 0,005 0,003 0,01 0,42 5C 35,7 77,7 59 0,46 0,605 1,32 24,03 SD 0,1 1,2 1 0,006 0,009 0,01 0,65 6C 37,4 85 66 0,44 0,567 1,29 22,36 SD 0,2 1 1 0,003 0,006 0 0,26 7C 35,5 81,7 64,7 0,434 0,548 1,26 20,82 SD 0,2 0,6 0,6 0,002 0,004 0 0,15 8C 36,6 84 67,3 0,435 0,543 1,25 19,84 SD 0,1 1 0,6 0,006 0,005 0,01 0,5 9C 36,2 80,3 66,7 0,451 0,544 1,21 17,02 SD 0,1 0,6 1,2 0,003 0,009 0,01 0,84 10C 36,6 77,3 60 0,474 0,611 1,29 22,39 SD 0,2 1,5 0 0,007 0,003 0,03 1,52 11C 37,1 81 64,3 0,458 0,577 1,26 20,57 SD 0,2 1 0,6 0,004 0,003 0,01 0,51 12C 36,1 77,7 63,3 0,464 0,569 1,23 18,45 SD 0,2 1,2 0,6 0,006 0,004 0,01 0,47 13C 40,1 86 76,3 0,467 0,526 1,13 11,25 SD 0,5 1,7 2,1 0,004 0,008 0,01 0,81 14C 37,3 80 68,3 0,466 0,545 1,17 14,57 SD 0,2 1 0,6 0,004 0,005 0,02 1,28 15C 38,7 79,7 68 0,485 0,569 1,17 14,65 SD 0,3 0,6 1 0,003 0,004 0,01 0,78 16C 39,4 81 70,7 0,486 0,557 1,15 12,75 SD 0,8 1,7 1,2 0,003 0,004 0,01 0,43 17C 38,9 84 74,7 0,463 0,521 1,12 11,11 SD 0,4 1 0,6 0,002 0,004 0,01 0,57 Flow character Passable Passable Passable Passable Passable Passable Passable Fair Fair Passable Passable Fair Good Good Good Good Good 63 Table 15. Pycnometric density of granules consisting of DH, MCC and NM 30D. Sample 1D 2D 3D 4D 5D 6D 7D 8D 9D 10D 11D 12D 13D Density g/cm3 1,4126 1,4099 1,3918 1,3911 1,3786 1,3727 1,3634 1,3633 1,3630 1,3539 1,3424 1,3384 1,3318 SD g/cm3 0,0002 0,003 0,0016 0,0016 0,0012 0,0015 0,0772 0,0229 0,0011 0,0003 0,0012 0,0011 0,0015 Table 16. Pycnometric density of granules consisting of C, MCC and NM 30D. Sample 1C 2C 3C 4C 5C 6C 7C 8C 9C 10C 11C 12C 13C 14C 15C 16C 17C Density g/cm3 1,4684 1,4624 1,4454 1,4357 1,4582 1,4507 1,4481 1,4345 1,4241 1,4347 1,4328 1,4266 1,4351 1,4229 1,4285 1,4192 1,4210 SD g/cm3 0,0026 0,0030 0,0013 0,0008 0,0013 0,0025 0,0016 0,0019 0,0024 0,0009 0,0037 0,0003 0,0015 0,21 0,0028 0,0030 0,038 64 Table 17. Granules consisting of DH, MCC and NM 30D size distribution. Sample 1D 2D 3D 4D 5D 6D 7D 8D 9D 10D 11D 12D 13D > 1,25 mm 0 0,1 0 0,1 0 0 0 0 0 0,1 0 0,1 0 1,25 1,0 mm 2,7 2,9 2,3 5,7 2,7 4,2 1,7 3,4 3,7 6,4 3,4 8,8 5,5 Size distribution of granules % 1,0 0,8 0,5 0,25 0,8 mm 0,5 mm 0,25 mm 0,125 mm 9 14,5 13,3 23,4 9,4 16,8 15,9 26,6 7,6 16,9 22 30,8 14,9 32,1 33,7 7,6 9,6 19 22,3 35,3 11,7 21,3 23,7 31 8,6 15,7 15,1 17,4 10,3 16,9 17,6 25,6 12 22,2 29,8 26,6 14,6 30,6 3,4 8,9 9,6 17,7 17,2 28,5 17,7 31,5 30 7,4 12,3 23 28,7 22,5 0,125 0,08 mm 17,8 17,9 14,8 5,3 8,8 6,6 26,6 19,4 4,1 4,7 19,7 4 6,9 < 0,08 mm 19,3 10,4 5,6 0,6 2,3 1,5 14,9 6,8 1,6 0,7 3,9 0,5 1,1 Table 18. Granules consisting of C, MCC and NM 30D size distribution. Sample 1C 2C 3C 4C 5C 6C 7C 8C 9C 10C 11C 12C 13C 14C 15C 16C 17C > 1,25 mm 0 0 0 0 0 0 0 0 0,1 0 0 0 0,1 0 0 0 0 1,25 1,0 mm 4,4 4,2 0,7 4,2 3 5,6 4,9 1,3 3,5 2,6 2,4 3,1 3,9 4,3 4,9 3,6 2,8 1,0 0,8 mm 8,8 9,2 5,7 8,1 6,8 7,8 8,9 7,2 7,9 6,3 8,2 9,4 11 11 11,3 14,5 11,9 Size of sieves mm 0,8 0,5 0,5 mm 0,25 mm 14,7 19 16,6 21,1 10,3 25,3 18,4 24,9 13,3 23,4 15,8 37,3 17,2 29,9 18,8 32,1 18,5 32,4 14,8 40 16,8 23,9 19,3 30,5 23,4 41,3 23,7 41 25,3 38,1 28 38,6 28,7 42,9 0,25 0,125 mm 33,6 32,7 29,7 27,6 46,8 29,7 35,1 32,5 27,5 29,3 27,9 27,4 18 18,1 18,7 14,5 13,1 0,125 0,08 mm 16,7 11,4 11,6 11,4 5,7 3,2 3,4 7,4 8,3 6,3 13,3 6,8 1,3 1,1 1 0,5 0,4 < 0,08 mm 2,8 4,8 7,7 5,4 1 0,6 0,6 0,7 1,8 0,7 7,6 3,5 0,9 0,8 0,7 0,3 0,2 65 ADDITION Nr. 3 Table 19. Mass and content uniformity of DH matrix tablet - minimal and maximal weight of tablets, allowed limits of weight deviation by Ph. Eur., average values of drug content. Sample 1D 2D 3D 4D 5D 6D 7D 8D 9D 10D 11D 12D 13D Minimal weight (mg) 211,23 215,16 216,51 233,78 241,97 247,38 159,79 164,48 167,38 172,96 126,62 134,88 138,68 Maximal weight (mg) 222,37 222,5 233,48 237,61 245,86 251,32 165,54 169,98 172,38 177,94 134,04 138,59 143,35 Limits of weight (mg) 200,88 - 233,46 205,54 - 238,87 210,89 - 245,09 217,11 - 252,31 223,34 - 259,56 229,56 - 266,78 147,58 - 171,52 150,64 - 175,06 154,16 - 179,16 158,18 - 183,84 122,98 - 142,92 125,56 - 145,92 128,50 - 149,34 Average DH content (mg) 111,03 109,17 105,27 102,3 103,54 102,4 95,12 96,21 98,42 98,42 100,4 98,04 101,94 SD (mg) 1,79 2,94 3,2 5,01 4,41 0,72 2,71 1,32 2,81 2,81 3,21 3,26 2,08 Table 20. Mass and content uniformity of C matrix tablet - minimal and maximal weight of tablets, allowed limits of weight deviation by Ph. Eur., average values of drug content. Sample 1C 2C 3C 4C 5C 6C 7C 8C 9C 10C 11C 12C 13C 14C 15C 16C 17C Minimal weight (mg) 214,8 220,8 226,8 234 159,5 161,7 165,7 170,8 175,2 132,9 134,3 138,4 141 143,7 148,1 150 155 Maximal weight (mg) 223,42 225,46 229,4 237,61 162,8 167,35 171,14 174,55 181,41 136,14 137,35 142,07 144,86 147,59 152,53 152,69 158,84 Limits of weight (mg) 201,37 - 234,03 205,54 - 238,87 210,89 - 245,09 217,11 - 252,31 147,58 - 171,52 150,64 - 175,06 154,16 - 179,16 158,18 - 183,84 162,86 - 189,26 122,98 - 142,92 125,56 - 145,92 128,49 - 149,33 131,81 - 153,19 132,39 - 153,85 135,71 - 157,71 139,60 - 162,24 143,52 - 166,79 Average C content (mg) 104,16 102,95 103,62 104,24 101,98 101,4 105,24 103,14 101,24 102,05 101,54 105,57 104,62 103,89 104,23 103,03 108,14 SD (mg) 3,55 3,06 4,58 1,12 1,67 3,66 3,17 2,68 1,93 3,4 3,02 2,94 4,41 3,88 5,87 2,22 5,09 66 Table 21. Friability of DH matrix tablets. Sample 1D 2D 3D 4D 5D 6D 7D 8D 9D 10D 11D 12D 13D Friability (%) 0,033 0,031 0,025 0,033 0,049 0,045 0,065 0,066 0,049 0,040 0,114 0,096 0,074 Table 22. Friability of C matrix tablets. Sample 1C 2C 3C 4C 5C 6C 7C 8C 9C 10C 11C 12C 13C 14C 15C 16C 17C Friability (%) 0,1 0,119 0,086 0,059 0,139 0,125 0,116 0,088 0,179 0,159 0,145 0,126 0,113 0,081 0,078 0,04 0,035 67 Figure 26. Hardness of DH matrix tablets 180 160 140 Hardness (N) 120 100 80 60 40 20 0 1D 2D 3D 4D 5D 6D 7D 8D 9D 10D 11D 12D 13D Sample Figure 27. Hardness of C matrix tablets. 160 140 Harness (N) 120 100 80 60 40 20 0 1C 2C 3C 4C 5C 6C 7C 8C 9C 10C 11C 12C 13C 14C 15C 16C 17C Sample 68 Figure 28. Press force used within preparation process of DH matrix tablets. 35 30 Force (kN) 25 20 15 10 5 0 1D 2D 3D 4D 5D 6D 7D 8D 9D 10D 11D 12D 13D Sample Figure 29. Press force used within preparation process of C matrix tablets. 25 Force (kN) 20 15 10 5 0 1C 2C 3C 4C 5C 6C 7C 8C 9C 10C 11C 12C 13C 14C 15C 16C 17C Sample 69 ADDITION Nr. 4 Figure 30. Dissolution profile of samples 1D, 2D and 3D in phosphate buffer of pH 6.8. 120 110 DH released amount (%) 100 90 80 70 60 Sample 1D 50 Sample 2D 40 Sample 3D 30 20 10 0 0 30 60 120 180 240 300 360 420 480 540 600 660 720 Time (min) Figure 31. Dissolution profile of samples 4D, 5D and 6D in phosphate buffer of pH 6.8. 100 90 DH released amount (%) 80 70 60 50 Sample 4D 40 Sample 5D 30 Sample 6D 20 10 0 0 30 60 120 180 240 300 360 420 480 540 600 660 720 Time (min) 70 Figure 32. Dissolution profile of samples 7D, 8D, 9D and 10D in phosphate buffer of pH 6.8. 110 DH released amount (%) 100 90 80 70 60 Sample 7D 50 Sample 8D 40 Sample 9D 30 Sample 10D 20 10 0 0 30 60 120 180 240 300 360 420 480 540 600 660 720 Time (min) Figure 33. Dissolution profile of samples 11D, 12D and 13D in phosphate buffer of pH 6.8. 100 90 DH released drug (%) 80 70 60 50 Sample 11D 40 Sample 12D 30 Sample 13D 20 10 0 0 30 60 120 180 240 300 360 420 480 540 600 660 720 Time (min) 71 Table 23. Standard deviation values of DH released amount from all samples within dissolution test. Standard deviation values of DH released amount within dissolution test (%) 1D 2D 3D 4D 5D 6D 7D 8D 9D 10D 11D 12D 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 10,74 0,96 1,06 2,35 2,59 1,30 2,67 1,84 0,96 1,28 9,04 1,02 14,47 0,91 1,07 2,76 3,42 1,92 2,35 1,52 1,26 1,38 10,94 6,51 11,56 1,93 2,22 1,73 1,08 3,32 2,97 5,57 0,14 4,23 5,24 3,23 3,98 2,43 3,75 0,74 2,16 4,59 2,31 13,54 0,59 4,92 0,76 3,83 5,23 0,94 4,43 0,41 3,97 5,15 1,32 13,34 1,32 5,02 0,79 3,66 4,46 1,97 4,58 2,67 5,86 5,45 1,34 12,56 1,47 4,88 0,85 3,69 3,95 2,12 4,85 3,21 4,81 5,61 1,54 11,50 1,43 4,70 0,84 3,76 3,76 3,10 5,05 3,2 5,23 5,70 1,54 10,40 1,57 4,47 0,78 3,73 3,65 3,39 5,35 3,16 4,48 5,71 1,38 9,18 1,67 4,22 0,65 3,38 3,45 3,66 5,66 3,05 4,95 5,78 1,32 8,01 1,88 3,97 0,61 2,83 3,26 3,82 6,25 2,94 5,86 5,77 1,18 6,78 1,99 3,73 0,53 2,17 3,08 3,85 6,77 2,71 5,55 5,79 1,02 5,65 2,17 3,43 0,44 1,60 3,05 3,93 6,97 2,42 6,29 5,67 0,95 4,52 2,17 3,17 0,37 1,14 Time (min) 0 30 60 120 180 240 300 360 420 480 540 600 660 720 13D 0,00 5,30 8,10 9,50 10,20 10,10 9,90 9,40 9,10 8,90 8,70 8,50 8,30 7,90 Figure 34. Dissolution profile of samples 1C, 2C, 3C and 4C in phosphate buffer of pH 6.8. 110 100 C released amount (%) 90 80 70 60 Sample 1C 50 Sample 2C 40 Sample 3C 30 Sample 4C 20 10 0 0 30 60 120 180 240 300 360 420 480 540 600 660 720 Time (min) 72 Figure 35. Dissolution profile of samples 5C, 6C and 7C in phosphate buffer of pH 6.8. 110 100 C released amount (%) 90 80 70 60 Sample 5C 50 Sample 6C 40 Sample 7C 30 20 10 0 0 30 60 120 180 240 300 360 420 480 540 600 660 720 Time (min) Figure 36. Dissolution profile of samples 8C and 9C in phosphate buffer of pH 6,8. 110 100 90 C released amount (%) 80 70 60 50 Sample 8C 40 Sample 9C 30 20 10 0 0 30 60 120 180 240 300 360 420 480 540 600 660 720 Time (min) 73 Figure 37. Dissolution profile of samples 10C, 11C, 12C and 13C in phosphate buffer of pH 6,8. 110 100 90 C released amout (%) 80 70 60 Sample 10C 50 Sample 11C 40 Sample 12C 30 Sample 13C 20 10 0 0 30 60 120 180 240 300 360 420 480 540 600 660 720 Time (min) Figure 38. Dissolution profile of samples 14C, 15C, 16C and 17C in phosphate buffer of pH 6,8. 110 100 90 C released amount (%) 80 70 60 Sample 14C 50 Sample 15C 40 Sample 16C Sample 17C 30 20 10 0 0 30 60 120 180 240 300 360 420 480 540 600 660 720 Time (min) 74 Table 23. Standard deviation values of C released amount from all samples within dissolution Standard deviation values of C released amount within dissolution test (%) test. Sample 1C 2C 3C 4C 5C 6C 7C 8C 9C 10C 11C 12C 13C 14C 15C 16C 17C 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 30 1,15 2,07 1,59 0,22 1,61 2,59 1,33 3,48 2,3 6,15 14,3 2,75 0,71 2,31 6,24 0,63 4,45 60 1,2 0,27 1,14 0,27 0,75 1,23 0,14 2,37 1,62 2,41 9,83 3,51 0,69 2,82 0,85 0,52 11,5 120 1,24 0,25 1,11 0,66 0,71 0,69 0,05 0,92 0,58 0,85 2,83 4,38 0,78 2,32 0,87 0,53 1,29 180 1,22 0,28 1,15 1,47 0,79 0,75 0,03 0,91 1,51 0,84 2,61 3,22 0,81 1,44 0,85 0,58 1,31 240 1,25 0,35 1,21 1,64 0,7 0,77 0,07 0,86 1,75 0,86 2,74 1,41 0,87 1,59 0,82 0,57 1,34 Time (min) 300 360 1,29 1,28 0,4 0,23 1,32 1,12 1,38 1,15 0,76 0,71 0,86 0,9 0,07 0,1 0,93 0,99 1,77 1,8 0,91 0,97 2,74 2,31 1,04 1,09 0,87 0,86 1,13 1,92 0,85 0,83 0,54 0,57 1,39 1,47 420 1,32 0,25 1,31 1,15 0,71 0,94 0,07 1,01 1,8 0,96 2,47 0,91 0,88 1,75 0,86 0,58 1,5 480 1,35 0,3 1,01 0,91 0,72 1,04 0,04 1 1,82 1,02 2,89 1,16 0,88 1,92 0,81 0,6 1,56 540 1,39 0,15 1,18 0,69 0,56 1,09 0,05 1,08 3,68 1,05 2,19 1,65 0,87 1,82 0,79 0,6 1,64 600 1,37 0,2 0,97 0,69 0,55 1,13 0,08 1,05 1,84 1,1 2,54 1,79 0,91 1,81 0,8 0,62 1,63 660 1,39 0,51 1,42 0,54 0,57 1,21 0,14 1,08 1,89 1,12 2,31 1,54 0,92 2,42 0,81 0,6 1,72 720 1,44 0,42 1,14 0,64 0,53 1,53 0,13 1,12 1,93 1,15 2,45 1,72 0,9 2,18 0,85 0,62 1,74 Figure 39. Dissolution profile of 14C matrix tablet within dissolution test with continual pH change (the first 2 h at pH 1.2 followed by 10 hours at pH 6,8). 100 90 C released amount (%) 80 70 60 50 40 30 20 10 0 0 30 60 120 180 240 300 360 420 480 540 600 660 720 Time (min) 75 Figure 40. Dissolution profile of 4D and 5D matrix tablets within dissolution test with continual pH change (the first 2 h at pH 1.2 followed by 10 hours at pH 6,8). 100 90 C released amount (%) 80 70 60 50 Sample 4D 40 Sample 5D 30 20 10 0 0 30 60 90 120 150 180 210 240 300 360 420 480 540 600 660 720 Time (min) Table 25. Standard deviation values of drug released amount from samples 4D, 5D and 14C within dissolution test with continual pH change. Time (min) 0 30 60 120 180 240 300 360 420 480 540 600 660 720 Standard deviation values of drug released amount within continual dissolution test (%) 4D 5D 14C 0,00 0,00 0,00 0,61 0,79 4,32 0,65 1,32 4,87 1,18 3,42 5,67 1,10 0,47 4,90 0,94 0,29 1,67 0,98 0,24 0,53 1,01 0,22 1,10 0,96 0,22 1,12 0,98 0,20 1,10 0,96 0,22 1,15 1,00 0,19 1,09 1,01 0,21 1,09 1,00 0,22 1,11 76 77