Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
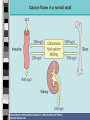
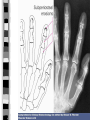
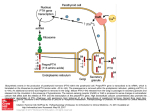
PARATHYROID GLAND DISEASES Primary hyperparathyroidism Hypoparathyroidism Causes of hypercalcemia Primary hyperparathyroidism: sporadic, associated with MEN 1 or MEN 2a, familial, after renal transplantation Secondary, tertiary hyperparathyroidism Malignancies: humoral hypercalcemia (caused by PTHrP, 1,25(OH)2D3, PTH), local osteolytic hypercalcemia Sarcoidosis Endocrinopathies: thyrotoxicosis, adrenal insufficiency, pheochromocytoma, acromegaly Drug induced: vitamin A, D intoxication, thiazides, lithium,milk-alkali syndrome, estrogens, androgens, tamoxifen Immobilization Acute renal failure P-HPTH Common, usually asymptomatic disorder 2-3fold commoner in females than in males Incidence approx. 42 per 100,000 inhibitants/year Single parathyroid adenoma – ca. 80%, parathyroid hyperplasia – ca. 15%, parathyroid carcinoma – 1-2% Defense mechanism against hypercalcemia Hypercalcaemia supression of PTH secretion bone resorption renal production of 1,25(OH)2D3 calcium resorption from intestine urinary calcium loss „Stones” „Bones” Renal stones Nephrocalcinosis Polyuria Polydipsia Uraemia Osteitis fibrosa with: subperiosteal resorption - osteoclastomas - bone cysts Radiologic „osteoporosis” Osteomalacia or rickets Arthrithis „Abdominal groans” Constipation Indigestion, nausea, vomiting Peptic ulcer Pancreatitis P-HPTH signs & symptoms Other Proximal muscle weakness Keratitis, conjunctivitis Hypertension Itching „Psychic moans” Lethargy, fatigue Depression Memory loss Psychoses – paranoia Personality change, neuroses Confusion, stupor, coma Hyperparathyroid bone disease Osteitis fibrosa cystica (< 10% of patients) Pain , pathologic fractures AlP Cystic lesions containing fibrous tissue („brown tumours”) or cyst fluid Subperiosteal resorption of cortical bone, „salt-and-pepper” appearance of the skull Secondary osteoporosis (loss of cortical bone) Hyperparathyroid kidney disease Kidney stones (< 15% of patients) Nephrocalcinosis Polidypsia, poliuria (loss of renal concentration ability) Gradual loss of renal function Other features of P-HPTH Lethargy, fatigue, depression, difficulty in concentrating, personality changes Frank psychosis Muscle weakness Hypertension Dyspepsia, nausea, constipation Chondrocalcinosis („pseudogout”), gouty arthritis Laboratory findings in P-HPTH total Cas (may be intermittent), Cau, Ps, Pu intactPTH (may be upper normal) hyperchloremic acidosis GFR Cas 2.25-2.75 mmol/l, Cau < 4 mg/kg/24 h (<250 mg/24 h in women, < 300 mg/24 h in men) Ps 3.0-4.5 mg% (1.0-1.5 mmol/l), Pu 400-1400 mg/24 h Treatment of P-HPTH The adenoma may be located throughout the neck or upper mediastinum „The only localization study needed in a patients with hyperparathyroidism is to locate an experienced parathyroid surgeon” Surgical parathyroidectomy cure rate over 95% (adenoma + excellent surgeon) Localization studies are very useful in reoperative parathyroid surgery: neck ultrasound, 99mTcsestamibi scanning, CT, MRI (rarely angiography, venous sampling) Treatment of P-HPTH (II) No definitive therapy for hyperparathyroidism Estrogen replacement therapy in postmenopausal women Management of hypercalcaemia: rehydrating with saline, furosemide, calcitonin s.c. (4-8 IU/kg every 12 hrs.), bisphosponates (etidronate disodium, pamidronate disodium), glucocorticoids p.o. (in multiple myeloma, sarcoidosis, intoxication with vitamin D or A). 1990 NIH Consensus Development Conference Surgery should be recommended if: 1) 2) 3) 4) 5) 6) 7) serum Ca is markedly elevated (above 2.8-3.0 mmol/l) if there has been a previous episode of lifethreatening hypercalcemia if creatinine clearance is reduced below 70% of normal if a kidney stone is present if urinary calcium is markedly elevated (> 400 mg/24 h) if bone mass is substantially reduced (less than 2 SD below normal for age, sex, and race) if the patient is young (under 50 years of age, particularly premenopausal women) Causes of hypocalcemia Hypoparathyroidism: surgical, idiopathic, neonatal, familial, postradiation, infiltrative Resistance to PTH action: pseudohypoparathyroidism, renal insufficiency, medications that block osteoclastic bone resorption (calcitonin, bisphosphonates) Failure to produce 1,25(OH)2D3: vitamin D deficiency, hereditary vitamin D-dependent rickets, type 1 (1-hydroxylase deficiency) Resistance to produce 1,25(OH)2D3: hereditary vitamin D-dependent rickets, type 2 (defective VDR) Acute complexation or deposition of calcium: acute hyperphosphatemia (crush injury, rapid tumour lysis, excessive enteral and parenteral phosphate administration), acute pancreatitis, citrated blood transfusion, hungry bones syndrome Clinical features of hypocalcemia Neuromuscular manifestations: overt tetany: carpopedal spasm, painful; laryngospasmus, blepharospasmus latent tetany: Chvostek’s sign, Trousseau’s sign focal or generalized seizures, papilledema, confusion, organic brain syndrome, mental retardation in children, calcification of basal ganglia (skull X-ray, CT) Cardiac effects: prolongation of QT interval, congestive heart failure Ophtalmologic effects: subcapsular cataract Dermatologic effects: dry and flaky skin, brittle nails, impetigo herpetiformis, pustular psoriasis Hypoparathyroidism Surgical, autoimmune, idiopathic, familial Cas, Ps, low or undetectable intact PTH level Surgical – ensues 1-2 days postoperatively, transient in 50% of cases Autoimmune – most commonly associated with Addison’s disease and mucocutaneous candidiasis (type I polyglandular autoimmune syndrome) Idiopathic – an isolated form, age of onset 2-10 years, commoner in females Familial – due to activating mutation of the parathyroid calcium receptor gene Pseudohypoparathyroidism (PHP) A heritable disorder of target-organ unresponsineness to PTH (Ellsworth-Howard test: lack of an increase in urinary cAMP after administration of exogenous PTH) Hypocalcemia and hyperphosphatemia, but elevated PTH level and a markedly blunted response to PTH administration 2 distinct forms: PHP 1A – characteristic somatic phenotype, i.e. Albright’s hereditary osteodystrophy (short stature, a round face, short neck, brachydactyly, subcutaneous ossifications) PHP 1B – no characteristic somatic phenotype Treatment of hypocalcemia Acute hypocalcemia: calcium chloride or gluconate i.v. (up to 400-1000 mg/24 h), oral calcium and vitamin D should be started (caution: digitalis treatment, stridor) Chronic hypocalcemia: objective: normalisation of serum calcium and phospate 1.0-2.0 g of elemental calcium p.o. per day, vitamin D3, active metabolites: alfacalcidol (1[OH]D3), calcitriol (1,25[OH]2D3), low phosphate diet (no milk)























![Poster ECE`14 PsedohipoPTH [Modo de compatibilidad]](http://s1.studyres.com/store/data/007957322_1-13955f29e92676d795b568b8e6827da6-150x150.png)





