Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
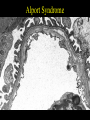
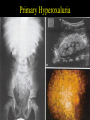
Inherited Renal Diseases Part II Maria Ferris and Deb Gipson 10/23/01 Outline • Lowe Syndrome • Fabry disease • Cystinosis • Cystinuria • Hyperoxaluria • Alports Fabry Disease • X-linked recessive disorder (Xq22-24) • Defect is in the alpha-1 galactosidase A • Diagnosis by assay of peripheral blood leukocyte level of alpha-galactosidase – Hemizygous(M) level is near 0 – Heterozygote (F) level may be in the low normal range. Check urinary ceramide digalactoside and trihexoside. – Fetus: determine level in amniocyte Fabry Disease • Males: wide range of clinical abnormalities • Females: variable range of clinical expression, may have lipid storage in cells and be completely asymptomatic (lyonization) • Race: White most common • Incidence: 1:40,000 • Intracellular accumulation of the glycosphingolipid galabiosylceramide Fabry: Deposition – *Endothelial, perithelial, smooth muscle of blood vessels – Kidney: glomerluar and tubular epithelium – Corneal epithelium – Myocardium – Autonomic NS: ganglion cells – RES: bone marrow, liver, spleen, lymph nodes – Lungs, synovial lining and testes Fabry: Clinical Manifestations – angiokeratoma (age 10-20) appearance dark red macules or papules – acroparesthesias exacerbated by fever and exercise (childhood) – anhidrosis (tears and saliva) – Nausea, abdominal pain, diarrhea – ophthalmic abnormalities: corneal opacity, posterior cataracts, – ischemic cerebrovascular disease: seizure, TIA, stroke – ischemic heart disease: MI, dysrhythmias Angiokeratoma Kidney – mild proteinuria (0.5-2g/d) in the 3rd decade – Uremia and hypertension in the 4-5th decade – ESRD as early as 2nd decade – Urine: myelin bodies, mild hematuria Pathology • Uniform-size empty vacuoles (formalin) – Most striking in visceral epithelial cells of glomeruli – Less in renal arterioles, tubular epithelium, mesangium, interstitium • Birefringence and Maltesecross w/ polarized optics (fresh or frozen) • Progressive disease: segmental and global glomerulosclerosis Pathology • IF is negative • EM: cellular inclusions in all glomerular cells – Zebra bodies or myelin figures – Onion skin appearance – Located in lysosomes • Foot process effusion w/ heavy proteinuria • DDX: – Pulmonary Scilicosis W/ hematuria and proteinuria: inclusions in glomeruli – Clororquine therapy: inclusions in glomeruli – Aminoglycoside: inclusions in tubules M&M in Fabry Disease • Natural Hx: death at mean age of 42 from uremia • Treated for ESRD: most common causes of death are cerebrovascular and cardiovascular • After transplant, the glycosphingolipid deposits recur but do not limit renal function • New Rx: IV alpha-galactosidase q 2 weeks. Approved in Europe last month. Awaiting FDA approval in US Alport Syndrome • Inheritance – Classic X-linked dominant – AR – AD • Gene: COL4A5 encodes for the alpha 5 chain of type IV collagen • Gene frequency 1:10,000 Alport Clinical Features • Hematuria: microscopic may be persistent, gross hematuria is intermittent if present. Onset 0-10 yrs • Proteinuria: absent in the first few years of life and then becomes gradually progressive. • Hypertension-progressive • Renal survival – Males: nearly all affected progress to ESRD; Age is variable – Females: better prognosis (X-linked variety); presence of gross hematuria in childhood, nephrotic syndrome, and diffuse GBM thickening are poor prognostic signs Alport Syndrome • Sensorineural hearing loss: onset by age 15 in males. Detect by audiometry. Progressive. In females progressive hearing loss is a poor prognostic sign • Ocular defects: 15-30% – Anterior lenticonus – Corneal endothelial vesicles • Platelet defects: megathrombocytopenia + platelet dysfunction (AD) • Diffuse leiomyomatosis: upper GI tract, tracheobronchial tree, females genital. Posterior subcapsular cataracts. (AD) Alport Nephropathology: Light – < 5 years: nearly nl w/ occasional interstitial foam cells and fetal glomeruli – mesangial widening – focal thickening of Bowman’s capsule – focal endothelial and mesangial proliferation – split capillary walls – progressive glomerular sclerosis – TBM thickening – interstitial fibrosis and foam cells; tubular atrophy – occasional crescents – capsular tuft synechiae Nephropathology • IF – Typically negative – Increased deposition w/ sclerosis • EM * Variable thickening, thinning, basket weaving, and lamellation of the GBM Alport Syndrome Alport: Prognosis • Post transplant – 5% Anti-GBM nephritis – responds to therapy – likely to recur in next allograft • Hearing deficit – may progress to total deafness • Treatment – supportive Primary Hereditary Hyperoxaluria • Type I – hyperoxaluria w/ glycolic aciduria – alanine: glyoxylate aminotransferase deficiency (peroxisome) – autosomal recessive; 1:60,000-120,000 • Type II – hyperoxaluria w/ L-glyceric aciduria – D-glycerate dehydrogenase deficiency (cytosol) – very rare • Type III – Intestinal hyperoxaluria (hyperabsorptive) (1)glycine cleavage enzyme; (2) alanine: glyoxylate aminotransferase; (3) Dglyceric acid dehydrogenase; (4) glycerate kinase; (5) trimethylamine oxidase; (6) lactate dehydrogenase; (7) glycolate oxidase; (8) NKH* = nonketotic hyperglycinemia; TH4 =tetrahydrofolate. Behrman: Nelson Textbook of Pediatrics, 16th ed. 2000 Metabolic Pathway Defective in PH-I glyoxalate Pyridoxine Alanine:glyoxylate aminotransferase (liver) oxalate glycine Primary Hyperoxaluria-I: Laboratory • Elevated urinary sodium, oxalate, glycolic acid, and glyoxylic acid • Laboratory Normal Values – Blood oxalate 10-140 mg/dl – Urine oxalate 10-40 mg/day – Oxalate/Creatinine urine • Infants < 0.3 mg/mg • 1-5 yo < 0.1 mg/mg • >5 yo < 0.05 mg/mg Pathology • Hyperoxalemia: wide spread deposition of oxalate = oxalosis • Renal deposition: nephrolithiasis, tubulointerstitial nephropathy, renal failure • Light Microscopy: calcium oxalate crystalline deposits in tubule, interstitium, interstitial fibrosis, late focal glomerular sclerosis. Stone in calyceal system are birefringent Extrarenal deposition • blood vessel walls • bone • bone marrow (myelopthesis) • joints • heart • spleen • liver • thymus • • • • • • • pituitary adrenal pancreas parathyroids thyroid brain heart (conduction defects) Primary Hyperoxaluria Primary Hyperoxaluria - I Treatment • Early diagnosis • Dietary restriction of glycine and high oxalate foods (spinach, rhubarb, beet roots, iced tea) • Pyridoxine • Hydration to limit stone formation (2.5 L/m2) • + Mg and phosphate supplementation • Liver / Kidney transplant