Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
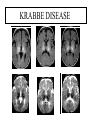
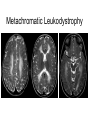
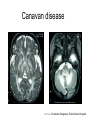
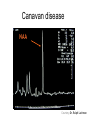
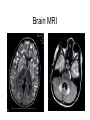
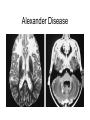
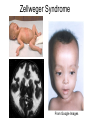
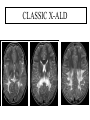
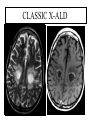
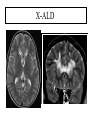
Leukodystrophy Tyler Reimschisel, MD September 6, 2013 Clinical Presentations of IMD • Intoxication – Urea cycle defects • Energy Failure – Mitochondrial disease – Glycogen storage disease • Complex Molecule – Lysosomal storage disease – Glycogen storage disease – Peroxisomal storage disorders Diseases that Cause Leukodystrophy Some examples • Adrenoleukodystrophy • Metachromatic leukodystrophy • Tay-Sachs • Krabbe • Canavan • Mitochondrial Clinical Presentation of Leukodystrophy • • • • • Developmental delay: relentless regression Seizures UMN signs Failure to thrive (less common) +/- dysmorphisms Testing for Leukodystrophy • • • • • Lysosomal enzyme profile VLCFA (very long chain fatty acids) Urine organic acids Lactate Pyruvate: not clinically useful lab due to timing; in equilibrium with alanine • Alanine (order via Plasma amino acids) Pelizaeus-Merzbacher • Xq22 mutation in proteolipid protein 1 (PLP1) • Onset in first few months of life with rotary head movements, rotary nystagmus, & motor delay • Then ataxia, tremor, choreoathetosis, spasticity • Seizures • Optic atrophy and ocular impairments • MRI: Reversal of gray-white signal due to diffuse dymyelination Pelizaeus-Merzbacher Gal-GalNAc Nana-Gal-Glc-Cer GM1 (- Galactosidase) GalNAc-Gal-Gal-Glc-Cer GalNAc Nana-Gal-Glc-Cer GM2 (-Hexosaminidase A) Nana-Gal-Glc-Cer Neuraminidase Sandhoff (-Hexosaminidase A & B) Gal-Gal-Glc-Cer Fabry (-Galactosidase) Gal-Glc-Cer Glc-Cer Gaucher (-Glucosidase) SO3H-Gal-Cer Gal-Cer Ceramide Phosphorylcholine-Cer (Sphingomyelin) Farber (Ceraminidase) Sphingosine Krabbe • AR defect of galactocerebroside-betagalactosidase on chromosome 14 • Pure neurologic condition • Onset at 3-8 months of age • Irritability, intermittent fevers, heightened startle reflex, feeding problems • Develop seizures, opisthotonus • Deafness and blindness by 9 months • MRI: KRABBE DISEASE Metachromatic Leukodystrophy • AR defect of arylsulfatase-A • Leukodystrophy as well as disease of adrenal glands, kidneys, pancreas, liver Metachromatic Leukodystrophy • 3 Presentations – Late infantile (18-24 months) • Gait disturbance, hypotonia to hypertonia, regression, involuntary movements, neuropathy, cherry red spot – Juvenile (4-10 years) • Bradykinesia, poor school performance, ataxia, movement disorder, neuropathy, slower progression – Adult • After puberty get personality and mental changes, cortical and cerebellar regression to frank dementia in third to fourth decade Metachromatic Leukodystrophy L.B. • 4-year-old girl with GDD, hypotonia, & worsening ataxia – Development at 12-18 month level – Hyperactivity, inattention and aggression (Tenex) • Family history – Maternal cousin with chromosome deletion – Paternal half-sister with B12 deficiency (?) • Labs – CMA, karyotype, FRX, purine/pyrimidines, biotinidase, MECP2, AS/PWS, EEG, brain MRI (9/2010) First Visit • Labs: PAA, acylcarnitine profile, vitamin B12, homocysteine, MMA level, creatine metabolites • Repeat brain MRI consistent with MLD Second Visit • Lysosomal enzyme panel, VLCFA, coenzyme Q10 level – Arylsulfatase A level 1.5 (low) – GM1, mannosidosis, fucosidosis, Krabbe, Tay-sachs normal Follow Up Testing • No mutations in arylsulfatase A gene • Parental testing showed normal arylsulfatase A enzyme activity Additional Testing • Arylsulfatase B enzyme activity at 4-5% normal • Huge peak of sulfatides in patient • Multiple sulfatase deficiency diagnosed • Molecular testing pending Multiple Sulfatase Deficiency • AR, mutations in sulfatase-modifying factor-1 gene (SUMF1) on 3p26 • Austria: 1 in 1.4 million individuals • Affects 12 sulfatase enzymes – Post-translation modification defect in which cystein residue of enzyme is not activated – Defect in enzyme that causes oxidation of a thiol group in cysteine to generate an alpha-formylglycine residue – Alpha-formylglycine residue may accept the sulfate during sulfate ester cleavage by hydrolysis – Examples: arylsulfatase, steroid sulfatase, heparan sulfatase, N-acetylglucosamine-6-sulfatase Multiple Sulfatase Deficiency • 3 phenotypes – Neonatal MSD: severe mucopolysaccharidosis – Late infantile MSD: late-onset MLD – Juvenile MSD • Combined features of MLD, Hunter, Sanfilippo A, Morquio, Maroteaux-Lamy, Xlinked ichthyosis Canavan • AR deficiency of asparto-acylase • Macrocephaly, lack of head control, and developmental delays by the age of three to five months • Develop severe hypotonia and failure to achieve independent sitting, ambulation, or speech • Hypotonia eventually changes to spasticity • Life expectancy is usually into the teens • Diagnosis of Canavan disease relies upon demonstration of very high concentration of N-acetyl aspartic acid (NAA) in the urine Canavan disease Courtesy Dr Isabelle Desguerre, Paris Necker Hospital Canavan disease NAA Courtesy Dr. Ralph Lachman L-2-Hydroxyglutaric Aciduria • Underlying defect unknown • Clinical – Normal to mild delays in infancy and early childhood – Slowly progressive encephalopathy – Variable rate of progressive ataxia, seizures, pyramidal signs, movement disorder (dystonia, tremor, choreoathetosis), dementia – 50% with macrocephaly • Laboratory: no metabolic decompensation, increased plasma lysine, elevated 2hydroxyglutaric acid in urine Brain MRI L-2-Hydroxyglutaric Aciduria • Neuroimaging – Severe cerebellar atrophy – Mildly swollen white matter with gyral effacement – Leukoencephalpathy more prominent closer to cerebral cortex – Increased signal intensity in dentate and striatum • Differential Diagnosis – D-2-hydroxyglutaric aciduria presents earlier – GAII causes elevations of D-2-hydroxyglutaric acid • Treatment - none Alexander Disease • AD mutation in GFAP at 17q21.31 • Onset at around 6 months (birth – 2 yrs) • Psychomotor regression, spasticity and seizures • Juvenile patients have ataxia and spasticity • Adult patients have MS-like presentation • Diffuse demyelination, especially in frontal lobes Alexander Disease Brain MRI: Leigh Syndrome From Osborn. Neuroradiology, 2000 Brain MRI From Osborn. Neuroradiology, 2000 Peroxisome Function • Synthesis – Plasmologens (ether-phospholipids) – Bile acid from mevalonate • Catabolism – -oxidize very long chain fatty acids (esp C24:0 and C26:0), pristanic acid and bile acid intermediates – -oxidize phytanic acid (chlorophyll derivative) to pristanic acid – Lysine via pipecolic acid and glutaric acid – Glyoxylate to prevent conversion to oxalate Peroxisomal Disorders • 16 disorders – 15 are autosomal recessive – 1 is X-linked (adrenoleukodystrophy) • Predominant features – Dysmorphisms – Neurologic dysfunction – Liver disease Peroxisomal Disorders • Biosynthesis Defects – Zellweger spectrum disorders (ZD, IRD, NR) – Rhizomelia chondrodysplasia punctata • Single Peroxisomal Enzyme Deficiencies – – – – – – – Adrenoleukodystrophy (ABCD1 on Xq28) RCDP type 2 (GNPAT on 1q42.1-42.3) RCDP type 3 (AGPS on 2q33) Refsum (PHYH/PAHX on 10p15-p14) Glutaric aciduria type 3 (?) Mulibrey nanism (TRIM on 17q22-23) 9 others Peroxisomal Biogenesis • Peroxisomes multiply by division • Proteins carried from free polyribosomes to peroxisomes by peroxisomal targeting signals (PTS) • PTS1 – Last 3 carboxy terminal amino acids – PTS1 receptor encoded by PEX5 • PTS2 – Stretch of 9 amino acids – PTS2 receptor encoded by PEX7 Peroxisomal Biogenesis • PTS receptors deliver proteins to peroxisomal protein import machinery • Import machinery transports proteins across membrane • Transporter complex has at least 15 peroxins (PEX1, 2, 3, 5, 6, 10, 12, 13, 14, 16, 19, 26) Zellweger Spectrum Disorders • CZ, NALD, and IRD • Genetic heterogeneity • Dysmorphism (large fontanelle, high forehead, abn ears, micrognathia, low/broad nose, redundant skin folds) • • • • • • • • Neuronal migration disorders and delayed myelination Seizures Hypotonia Sensorineural deafness Ocular abnormalities (retinopathy, cataracts, ON atrophy) Liver disease (hepatomegaly, cholestasis, hyperbilirubinemia) Failure to thrive Death in first year of life Zellweger Syndrome From Google Images ZELLWEGER SYNDROME Zellweger Spectrum Disorders • Classic Zellweger (CZ) • Neonatal adrenoleukodystrophy (NALD) – Somewhat less severe than CZ – May lack dysmorphisms altogether – Neonatal or infantile onset of seizures, hypotonia, and progressive leukodystrophy – May have pachypolymicrogyria • Infantile Refsum disease (IRD) – – – – – – Least severe phenotype, regression over time May be asymptomatic at birth No progressive leukodystrophy Variable expressivity of cognitive dysfunction Deafness and vision changes (retinopathy) May survive to adulthood Adrenoleukodystrophy/ Adrenomyeloneuropathy • Most common peroxisomal disorder (1/20,000) • Mutation in ABCD on Xq28 leads to defect in peroxisomal uptake of VLCFA • ALD: progressive neurologic disorder that begins at 512 years – – – – – Boys with new onset school difficulties & ADHD Visuo-spatial deficits and hearing loss Spasticity, ataxia, maybe seizures Hypoglycemia, salt losing, hyperpigmentation Rx: steroids, presymptomatic stem cell transplant, Lorenzo’s oil ineffective (oleic and erucic acids) • AMN: early adulthood progressive spastic paraparesis, cerebral demyelination (males) CLASSIC X-ALD CLASSIC X-ALD X-ALD