Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
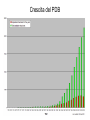
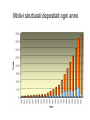
Dalla sequenza alla struttura Mauro Fasano Dipartimento di Biologia Strutturale e Funzionale Centro di Neuroscienze Università dell’Insubria – Busto Arsizio [email protected] http://fisio.dipbsf.uninsubria.it/cns/fasano Dalla sequenza alla struttura V L S E G E W Q L V L V . . . Sequenza O2 Struttura Funzione Che informazioni offre la struttura? • • • • • • Conformazione dei siti attivi e di legame Orientazione dei residui conservati Interpretazione di meccanismi Visualizzazione di cavità Calcolo di potenziale elettrostatico … Esempio • FtsZ – divisione cellulare in procarioti, mitocondri e cloroplasti. • Tubulina – componente strutturale dei microtubuli – comunicazione intracellulare e divisione cellulare. • FtsZ e Tubulina hanno bassa similarità di sequenza e non sembrerebbero omologhe. Burns, R., Nature 391:121-123 Picture from E. Nogales FtsZ e tubulina sono omologhe? • Proteine che hanno conservato la struttura tridimensionale possono derivare da un progenitore comune anche se la divergenza della sequenza non permette più di riconoscere l’omologia. Un altro esempio • α-lattalbumina e lisozima possiedono: – Stesso fold – Moderata similarità – Diversa funzione Metodi sperimentali: • Diffrazione dei raggi x • Risonanza magnetica nucleare Cristallografia a raggi X • Ottenere cristalli della proteina – 0.3-1.0 mm – Le singole molecole sono ordinate in modo periodico, ripetitivo. • La struttura è determinata dai dati di diffrazione. Image from http://www-structure.llnl.gov/Xray/101index.html Schmid, M. Trends in Microbiology, 10:s27-s31. Cristallografia a raggi X • Le proteine devono cristallizzare – Grande quantità – Solubili • Accesso a radiazione adatta • Tempo di calcolo per risolvere la struttura Risonanza Magnetica Nucleare (NMR) • • • • • • Proteine in soluzione Limite di dimensione ~ 40 kDa Proteine stabili a lungo Marcatura con 15N, 13C, 2H. Strumentazione molto costosa Tempo per assegnare le risonanze Il Protein Data Bank Crescita del PDB Motivi strutturali depositati ogni anno Percentuale di nuovi motivi strutturali HEADER COMPND COMPND COMPND COMPND COMPND COMPND SOURCE SOURCE SOURCE SOURCE KEYWDS EXPDTA AUTHOR REVDAT JRNL JRNL JRNL JRNL JRNL JRNL REMARK BINDING PROTEIN 01-JUN-95 1HXN MOL_ID: 1; 2 MOLECULE: HEMOPEXIN; 3 CHAIN: NULL; 4 DOMAIN: C-TERMINAL DOMAIN; 5 SYNONYM: HPX; 6 HETEROGEN: PO4 MOL_ID: 1; 2 ORGANISM_SCIENTIFIC: ORYCTOLAGUS CUNICULUS; 3 ORGANISM_COMMON: RABBIT; 4 TISSUE: SERUM HEME X-RAY DIFFRACTION H.R.FABER,E.N.BAKER 1 15-OCT-95 1HXN 0 AUTH H.R.FABER,C.R.GROOM,H.BAKER,W.MORGAN,A.SMITH, AUTH 2 E.N.BAKER TITL 1.8 ANGSTROMS CRYSTAL STRUCTURE OF THE C-TERMINAL TITL 2 DOMAIN OF RABBIT SERUM HEMOPEXIN REF TO BE PUBLISHED REFN 0353 1 1HXN 1HXN 1HXN 1HXN 1HXN 1HXN 1HXN 1HXN 1HXN 1HXN 1HXN 1HXN 1HXN 1HXN 1HXN 1HXN 1HXN 1HXN 1HXN 1HXN 1HXN 1HXN 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 ATOM ATOM ATOM ATOM ATOM ATOM ATOM ATOM ATOM ATOM ATOM ATOM ATOM ATOM ATOM ATOM 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 CA C O N CA C O CB OG N CA C O CB OG1 CG2 GLU GLU GLU SER SER SER SER SER SER THR THR THR THR THR THR THR 225 225 225 226 226 226 226 226 226 227 227 227 227 227 227 227 -0.900 -0.185 -0.514 0.788 1.534 2.231 1.883 2.572 3.237 3.242 3.989 4.274 4.179 5.354 5.114 6.256 -1.002 0.146 1.329 -0.203 0.805 1.806 1.952 0.130 -0.941 2.478 3.417 2.705 3.296 3.797 4.682 4.492 39.233 39.970 39.758 40.823 41.594 40.681 39.514 42.515 41.848 41.223 40.410 39.080 38.022 41.074 42.172 40.065 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 70.00 70.00 70.00 70.00 70.00 68.89 70.00 70.00 70.00 65.51 70.00 56.25 44.63 70.00 70.00 70.00 1HXN 1HXN 1HXN 1HXN 1HXN 1HXN 1HXN 1HXN 1HXN 1HXN 1HXN 1HXN 1HXN 1HXN 1HXN 1HXN 170 171 172 173 174 175 176 177 178 179 180 181 182 183 184 185 http://www.expasy.ch/spdbv Esegui Classificazione delle proteine: • SCOP (Structural Classification of Proteins, scop.mrc-lmb.cam.ac.uk/scop/, Murzin et. al.): 548 folds (major structural similarity in terms of secondary structures e.g. globin-like, Rossman fold); 1296 families (clear evolutionary relationship or homology e.g. globins, Ras) • CATH (Class, Architecture, Topology, Homologous Superfamily, www.biochem.ucl.ac.uk/bsm/cath/, Orengo et. al): 35 architectures (gross arrangment of secondary structures e.g. non-bundle, sandwich); 580 topologies (connectivity of secondary structures e.g. globin-like, Rossman fold); 1846 families (clear homology, same function) Structural Classification Of Proteins Predizione della struttura secondaria e terziaria Metodi predittivi »Comparative modeling > 30% similitudine »Threading/Fold recognition 0 – 30% similitudine »Ab initio nessun omologo Qualità del modello comparativo Identità di sequenza: 60-100% Confrontabile con NMR media risoluzione Specificità di substrato 30-60% Molecular replacement in cristallografia Partenza per site-directed mutagenesis <30% Gravi errori Building by homology (Homology modelling) Allineamento con proteine a struttura nota M A A G Y A Y G V L S - A T G F D - - V I D - A S G F E - - V V E - A K A Y L - - V L S Modello strutturale Fold recognition (Threading) Sequenza: MA A G Y AV L S + Motivi strutturali noti Modello strutturale Ab initio Sequenza MA A G Y AV L S Modello strutturale General Flowchart Building by homology Un numero grandissimo di polipeptidi si struttura in un numero finito (e relativamente piccolo) di folds Almeno una proteina su due di quelle presenti nel database ha un omologo (identità > 30%) che quasi sempre ha lo stesso fold. Costruire il modello comparativo 1) Cercare il massimo numero di omologhi che possiedano una entry nel PDB. Strumenti che utilizzano PSSM sono più sensibili. In questo caso vengono utilizzate sequenze senza struttura per costruire la PSSM. 2) Costruire un accurato allineamento multiplo tra la sequenza da modellare e tutte le entries che verranno utilizzate come templato. Trovare strutture di proteine la cui sequenza è simile allineamento Modello strutturale Verifica OK! Costruire il modello stesso Determinare la struttura secondaria in base all’allineamento Costruire le regioni conservate. Per ciascuna regione possiamo prendere le coordinate del frammento con la maggior similarità di sequenza. Costruire le regioni variabili, solitamente loops. Costruzione dei loops: Usando raccolte di loops osservati in strutture note, in base alla loro lunghezza ed alla loro sequenza Costruendo la conformazione del loop ab initio. Vengono generate numerose conformazioni casuali e si calcola l’energia in un opportuno campo di forze. Alcuni siti web di homology modeling COMPOSER – felix.bioccam.ac.uk/soft-base.html MODELLER – guitar.rockefeller.edu/modeller/modeller.html WHAT IF – www.sander.embl-heidelberg.de/whatif/ SWISS-MODEL – www.expasy.ch/SWISS-MODEL.html Swiss-Model http://www.expasy.ch/swissmod/SWISS-MODEL.html Modeller http://guitar.rockefeller.edu/modeller/about_modeller.shtml Advanced program for homology modeling Based on distance constraints Implemented in several popular modelling packages such as InsightII The source is available for unix platforms at the above URL Threading (fold recognition) La sequenza di input viene confrontata con una libreria di folds noti Si calcola un punteggio che esprima la compatibilità tra la sequenza e ciascun fold considerato Punteggi statisticamente significativi indicano che la sequenza ha una certa probabilità di assumere la stessa struttura 3D del fold considerato Input: Sequenza Donatore H Accettore H Gly Idrofobico Collezione di folds di proteine note Input: Sequenza Donatore H Accettore H Gly Idrofobico Collezione di folds di proteine note Donatore H Accettore H Gly Idrofobico S=-2 Z= -1 S=5 Z=1.5 S=20 Z=5 Chain/Domain Library Scoring functions for fold recognition Ci sono due metodi per valutare la compatibilità sequenzastruttura (1D-3D) In methods based on structural profile, for every fold a profile is built based on structural features of the fold and compatability of every amino acid to the features. The structural features of each position are determined based on the combination of secondary structure, solvent accessibility and the property of the local environment (hydrophobic/hydrophilic) The profile is a defined mathematical structure, adjusted for pair-wise comparisons and dynamic programming Amino acid type Position on sequence A 1 N D … 10 -50 101 2 -24 : C : 87 -99 : : : Y Gop Gext -80 100 10 167 100 10 : : : 100 10 Contact potentials This method is based on predefined tables which include pseudo-energetic scores to each pairwise interaction of two amino acids. This method makes use of distance matrix for representation of different folds For each pair of amino acids which are close in space the interaction energy is summed. The total sum is the indication for the fitness of the sequence into that structure Scoring Function …YKLILNGKTKGETTTEAVDAATAEKVFQYANDNGVDGEW… Tendenza a stare vicini: E_p Qualità dell’allineamento in una certa posizione: E_m (pairwise term) (mutation term) Alignment gap penalty: E_g Tendenza a stare in un certo ambiente: E_s (singleton term) Energia totale: E_m + E_p + E_s + E_g Descrive quanto la sequenza assomiglia al templato Amino acid index 1 • •• • N 1 • •• • • • •• • • • • •• • • • •• • •• N Amino acid index Expected Performance PROSPECT prediction in CASP4: 12 out 19 folds (no homology) recognized Predicted model X-ray structure Web sites for fold recognition Profiles: 3D-PSSM - http://www.bmm.icnet.uk/~3dpssm Libra I - http://www.ddbj.nig.ac.jp/htmls/E-mail/libra/LIBRA_I.html UCLA DOE - http://www.doe-mbi.ucla.edu/people/frsvr/frsvr.html Contact potentials 123D - http://www-Immb.ncifcrf.gov/~nicka/123D.html Profit - http://lore.came.sbg.ac.at/home.html Risultati Ab initio methods for modelling This field is of great theoretical interest but, so far, of very little practical applications. Here there is no use of sequence alignments and no direct use of known structures The basic idea is to build empirical function that simulates real physical forces and potentials of chemical contacts If we will have perfect function and we will be able to scan all the possible conformations, then we will be able to detect the correct fold Algorithms for Ab initio prediction include: A. Searching procedure that scans many possible structures (conformations) B. Scoring function to evaluate and rank the structures Due to the large search space, heuristic methods are usually applied The parameters in the searching procedure are the dihedral angles which specify the exact fold of the polypeptide chain A B C D E A B C D E New Fold Methods • Since almost all predictors use sequence and structural databases in some form, there is no longer an “ab initio” category • Assessment is sometimes difficult to communicate due to the complexity of the protein structure and completeness of prediction • Methods are still somewhat limited to smaller proteins Rosetta-David Baker • Based on the assumption that the distribution of conformations sampled by a local segment of the polypeptide chain is reasonably approximated by the distribution of structures adopted by that sequence and closely related sequences in known protein structures. • Fragment libraries for all possible three and nine residue segments of the chain are extracted from PDB by profile methods Rosetta-Simulation Procedure • Information on fragments from secondary structure prediction methods compiled and scored based on equation for local secondary structure propensity • Conformational space defined by these fragments is then searched by a Monte Carlo procedure with an energy function that favors compact structures with paired beta strands and buried hydrophobic residues, refinement of procedure late in simulation • Thousands of structures generated • Filters to remove bad structures • Remaning structures clustered and cluster center taken as the prediction. Methods to evaluate structures are based on Force fields- collection of terms that simulate the forces act between atoms Terms based on probabilities to find pairs of amino acids or atoms within specific distances Terms based on surface area and overlapping volume of spheres representing atoms Side chain construction In homology modelling, construction of the side chains is done using the template structures when there is high similarity between the built protein and the templates Without such similarity the construction can be done using rotamer libraries A compromise between the probability of the rotamer and its fitness in specific position determines the score. Comparing the scores of all the rotamer for a given amino acid determines the preferred rotamer. In spite of the huge size of the problem (because each side chain influences its neighbours) there are quite succesful algorithms to this problem. In this work we examined differences in structures of amino- acid side chains around point mutations. Conformation - a given set of dihedral angle which defines a structure. Asn Rotamer - energetically favourable conformation. Phe Example to library of rotamers SER 59.6 41.0 SER -62.5 26.4 SER 179.6 32.6 TYR 63.6 90.5 TYR 68.5 -89.6 TYR 170.7 97.8 TYR -175.0 -100.7 TYR -60.1 96.6 TYR -63.0 -101.6 21.0 16.4 13.3 20.0 10.0 19.3 Model evaluation After the model is built we can check it by various methods. If the model turns out to be bad, it is necessary to repeat several stages of the model building The main approaches for model evaluation are: A. Use of internal information (such as the one that used for the model construction) B. Use of external information derived from the databases Usually algorithms are checked by building models for proteins which have already solved structure and comparison between the model and the native structure It is always possible that information from the native structure will be used in direct or indirect ways for model building A more objective test is prediction of structures before they are publicly distributed (this is the idea of the CASP competitions)