Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
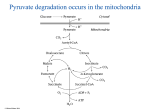
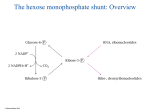
Metabolic uses of amino acids ● building blocks for protein synthesis ● precursors of nucleotides and heme ● source of energy ● neurotransmitters ● precursors of neurotransmitters and hormones Outline of amino acid degradation ● The liver is the major site of degradation for most amino acids, but muscle and kidney dominate the degradation of specific ones ● Nitrogen is removed from the carbon skeleton and transferred to α-ketoglutarate, which yields glutamate ● The carbon skeletons are converted to intermediates of the mainstream carbon oxidation pathways via specific adapter pathways ● Surplus nitrogen is removed from glutamate, incorporated into urea, and excreted Amino acid breakdown pathways join mainstream carbon utilization at different points of entry © Michael Palmer 2014 Transamination of amino acids © Michael Palmer 2014 The reaction mechanism of transamination © Michael Palmer 2014 The ping pong bi bi mechanism of transamination © Michael Palmer 2014 Nitrogen disposal and excretion ● Nitrogen accruing outside the liver is transported to the liver as glutamine or alanine ● In the liver, nitrogen is released as free ammonia ● Ammonia is incorporated into urea ● Urea is released from the liver into the bloodstream and excreted through the kidneys The urea cycle, part 1: carbamoylphosphate synthetase © Michael Palmer 2014 The urea cycle, part 2: subsequent reactions © Michael Palmer 2014 The urea cycle in context © Michael Palmer 2014 The urea cycle spans mitochondria and cytosol © Michael Palmer 2014 The glucose-alanine cycle © Michael Palmer 2014 Nitrogen transport by glutamine © Michael Palmer 2014 The central role of glutamate in nitrogen disposal © Michael Palmer 2014 Control of ammonia levels in the liver lobule © Michael Palmer 2014 Regulation of the urea cycle © Michael Palmer 2014 Hereditary enzyme defects in the urea cycle ● may affect any of the enzymes in the cycle ● urea cannot be synthesized, nitrogen disposal is disrupted ● ammonia accumulates, as do other metabolites depending on the deficient enzyme ● treatment ○ protein-limited diet ○ arginine substitution ○ alternate pathway therapy Asparagine degradation © Michael Palmer 2014 Serine dehydratase © Michael Palmer 2014 Serine-pyruvate transaminase © Michael Palmer 2014 Degradation of leucine © Michael Palmer 2014 Degradation of phenylalanine and tyrosine © Michael Palmer 2014 Phenylketonuria (PKU) ● homozygous defect of phenylalanine hydroxylase ● affects one in 10,000 newborns among Caucasians; frequency differs with race ● excess of phenylalanine causes symptoms only after birth; intrauterine development normal ● cognitive and neurological deficits, probably due to cerebral serotonin deficit ● treatment with phenylalanine-restricted diet ● some cases are due to reduced affinity of enzyme for cofactor THB, can be treated with high dosages of THB The Guthrie test for diagnosing phenylketonuria © Michael Palmer 2014 Ochratoxin A inhibits phenylalanyl-tRNA synthetase © Michael Palmer 2014 Tyrosinemia ● homozygous defect of fumarylacetoacetate hydrolase ● fumarylacetoacetate and preceding metabolites back up ● fumaryl- and maleylacetoacetate react with glutathione and other nucleophiles, causing liver toxicity ● the drug NTCB inhibits p-hydroxyphenylpyruvate dioxygenase, intercepting the degradative pathway upstream of the toxic metabolites ● dietary restriction of tyrosine required to prevent neurological deficit