Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
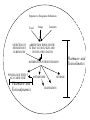
Toxicokinetics 1 Crispin Pierce, Ph.D. University of Washington [email protected] (206) 616-4390 Exposure to Exogenous Substances Food SECRETION OF ENDOGENOUS SUBSTANCES Drugs Toxicants ABSORPTION THROUGH THE GI TRACT, LUNGS, SKIN AND VENOUS CIRCULATION DISTRIBUTION WITHIN THE BODY PHYSIOLOGIC EFFECT AT A TARGET SITE Pharmaco- and Toxicodynamics METABOLISM STORAGE ELIMINATION Pharmaco- and Toxicokinetics Absorption Absorption assumes primary importance in oral, inhalation, and dermal exposures. The two kinetic parameters of concern are the rate of absorption and the extent of absorption (or bioavailability). Rate of Absorption The rate of absorption determines the time of onset and the degree of acute toxicity. This is largely because time to peak (Tpeak) and maximum concentration (Cmax) after each exposure depend on the rate of absorption. Rate the following processes in order of fastest to slowest: ORAL, DERMAL, INHALATION, INTRAVENOUS EXPOSURE. A C-max Conc. minimum toxic conc. B T-peak Slowing of absorption (AB) - prolonged Tp - lower Cmax Time In instances when the absorption rate is slower than elimination rate, the rate of washout of toxicant becomes rate-limited by absorption rather than by elimination (i.e., a depot effect). Absorption faster than elimination Elimination faster than absorption i.v. dose i.v. dose log Conc. non-i.v. dose log Conc. non-i.v. dose Time Time ? How does having pizza with your beer get you drunk more slowly? Systemic Availability The actual extent of exposure as defined by the amount of toxicant reaching the systemic circulation is determined by (1) entry barrier permeability, and (2) the extent of "first-pass" metabolism. The fraction of dose reaching the system circulation in intact form, or systemic availability (F), is estimated from either the AUCs, F = (AUCroute/AUCi.v.) Or from the amount of intact toxicant excreted in urine or exhaled via the lungs (Aex). F = (Aex-route/Aex-i.v.) ? Is mercury amalgam in tooth fillings dangerous? Modeling Absorption Intravenous dosing IVrate = IVdose / Timeinf and input into venous blood. Percutaneous dosing Percrate = (Percdoseexp(-KA,percTime))KA,perc and input into venous blood Oral dosing Oralrate = (Oraldoseexp(-KAoralTime)) KA oral and input into liver Inhalation dosing Inhalationrate = CartQc Qp * (Cinh- Calv) = Qc * (Cart - Cven) kblood/air = Pblood/air = Cart / Calv Cart = (QpPb/aCinh + CvenPb/aQc)/(QcPb/a + Qp) Volume of Distribution The Volume of Distribution is the apparent volume into which a drug or toxicant distributes, and provides a proportionality constant between blood (or plasma) concentration and the amount in the body: Volume of Distribution = Amount / Concentration Co Ln of Blood (or Plasma) Conc. The volume of distribution can be readily calculated V = Dose / Co after an intravenous bolus dose of a substance that exhibits "one-compartment model" characteristics: Time Volume of Distribution = Dose / Initial Concentration V = Dose / k•AUC Ln of Blood (or Plasma) Conc. slope = -k AUC Time However, because of the uncertainty in the estimate of Co, volume can be more accurately estimated by V = Dose / (kAUC), where AUC is the area under the concentration-time curve. The volume of distribution does not necessarily correspond to any physiologic volume, and is influenced by binding to plasma and tissue constituents. Volume can range from about 3 liters (as is seen with Tolbutamide, which is distributed in blood only, to about 50,000 liters (as is seen with Quinacrine, which distributes and binds to many tissues). The volume of distribution relates blood conc. to the total body burden of a toxicant, i.e., ABody = VCblood Physiologic Meaning? A measure of extravascular distribution. Two determinants of distribution into a tissue region: Tissue or organ volume Vti Distribution or Partition ratio Ptissue/blood = Cti/Cblood a constant @ pseudo-distribution equilibrium or steady state. Accordingly, ith Tissue Load = VtiCti = Vti(Pi,Cblood) Total Tissue Load = VtiPiCblood Total Body Load = Amount in blood + Amount in tissues ABody = VbloodCblood + VtiPiCblood = (Vblood + VtiPi)Cblood V = ABody/Cblood = Vblood + VtiPi where Vblood, Vti and Pi are constant. Since Pi can assume a value ~0-, V varies from a minimum of Vblood to many times the body size. Because the volume of distribution reflects the degree of xenobiotic dispersal and binding to all tissues, the following relationship is observed:Vinitial< Vsteady-state< Vterminal phase ? Would a chemical that is highly soluble in water, such as ethanol, have a large or small volume of distribution? How about a chemical that is highly soluble in fat, such as dioxin (TCDD)? Clearance Clearance is a measure of the body's ability to completely clear a drug or toxicant from blood or plasma. Clearance is the rate of elimination by all routes relative to the concentration in a systemic biologic tissue, and is measured in units of flow, or volume per unit time. CL (units of volume/time) = Rate of elimination (units of mass/time) / Concentration (units of mass/volume) Clearance is normally measured by collecting blood concentration-time data following a known dose, and using the following equation: CL (units of volume/time) = F*Dose (units of mass) / AUC (units of time-mass/volume) where F is the bioavailability (fraction of dose entering systemic circulation), and AUC is the area under the blood concentrationtime curve. Blood (or plasma) Concentration CL = F·Dose/AUC AUC Time Clearance also plays a role in determining the steady-state concentration of a drug or toxicant: Csteady-state = Rate of administration/ CL Area Under the Blood Concentration Time Curve (AUC): an internal or systemic exposure index. AUC o Cb dt o C0 • e dt C0 / k kt & since C0 = Dose/V, then AUC = Dose/kV The product kV is equal to clearance. AUC = Dose/CL or CL = Dose/AUC i.e., clearance governs the extent of systemic exposure as represented by AUC for a given dose of toxicant. Physiologic Basis of Clearance Blood clearance can be resolved into components representing the various metabolic and excretory pathways of elimination, e.g., CL = CLmetabolism + Clexhalation or further resolved into organ clearances, e.g., CL = (CLliver + CLg.i. tract + CLkidney + CLlung + ...) Individual organ clearance can in turn be related to organ blood flow (Qi) and extraction efficiency (Ei). For instance, Hepatic Clearance (CLliver) = QliverEh, note that Eh varies from 0–1 (i.e., 0 to 100% extraction) ? Would rapid breathing increase the clearance of a substance that leaves the body through the breath (such as nitrous oxide used in dentistry)? Half-Life Ln of Blood (or Plasma) Conc. C 1/2 C Half-life Time The Half-life is a measure of how rapidly a steady-state concentration will be achieved during constant rate dosing, and conversely how rapidly the concentration will fall after cessation of exposure. Half-life is related to the elimination rate constant k by the formula: t1/2 = ln 2 / k The elimination rate constant, like the clearance, is a fractional rate of decline: k = Rate of elimination / Amount Since CL = Rate of elimination/Concentration, the elimination rate constant can be estimated: k = CL / Volume of Distribution Half-life can then be found by t1/2 = ln 2/ k = ln 2 * V / CL Elimination Half-life (t0.5, t1/2) is a characteristic of First-order kinetics. For a one-compartment model: dABody= - k ABody Since ABody declines with time, elimination rate also decreases! However, the fractional rate is a constant, i.e., - 1 ABody -dABody/ABody —— ——— ——————— = k (time-1) ABody dt dt Upon integration, ABody = A0e-kt A0 = Body load @ t=0 But blood conc. rather than body load is measured. ABody ~ Cblood and ABody = VCblood Cblood = C0e-kt or Ln Cblood = Ln C0 kt where C0=Blood conc. @ t=0 Note that when Cblood = 1/2C0, t = 0.693/k = t1/2 It always takes 1 t1/2 to reach 50% of any starting conc. (i.e., t1/2 independent of C0) Takes about 3-4 t1/2s to effect 90% of elimination or to achieve 90% of the steady-state value under constant exposure. For compounds with multicompartmental kinetics, there will be a t1/2 estimate for each of the exponential phases. The terminal t1/2 is often quoted as the "Elimination t1/2," whereas the t1/2s of the earlier phases are referred to as "Distribution t1/2s.” For example, in a two compartment model described by Cblood = Ae-at + Be-bt , t1/2,a = 0.693/a and t1/2,b = 0.693/b ? How would half-life be affected if a condition such as kidney failure doubled the volume of distribution for a particular drug? Does drinking coffee or another source of caffeine help you to sober up? (Hint: caffeine does not affect the volume of distribution or clearance of ethanol.) Why are certain subpopulations (e.g., pregnant women, children) more susceptible to methyl mercury toxicity? (Hint: Might certain populations get higher doses of chemicals, possibly concentrated in smaller masses of tissue?)