Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
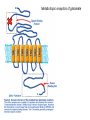
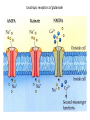
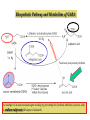
IL SISTEMA NEURONALE GABA/GLUTAMMATO Different Types of NTs 1. Amine a. 2. Acetylcholine (Ach) Monoamines a. b. Catecholamines i. Dopamine ii. Norepinephrine Indoleamines i. Serotonin 3. Amino acids 4. a. GUTAMMATO b. Aspartate c. GABA d. Glycine Peptides Amino Acid Neurotransmitters • Inhibitory – GABA and Glycine – Hyperpolarize = don’t fire • Excitatory – Glutamate ( and Aspartate) – Depolarize = fire 3 Deux types de neurones Pyramidal cells GLUTAMATE Excitation Interneurones GABA Inhibition GABA is a Gamma-aminobutyric acid Major inhibitory NT of the brain Usually involved with the transmission from one part of the CNS to the another Most CNS neurons have GABA receptors Neurotransmission by g-aminobutyric acid (GABA) is a major inhibitory mechanism in the human central nervous system (CNS). Postsynaptic GABAA ion channels open in response to the binding of GABA, and the ensuing chloride flux hyperpolarizes the membrane of the postsynaptic neurone inhibiting further neuronal activity. Gabaergic and Glutamatergic pathways in the CNS Endocrine role of GABA There are 3 major classes of GABA receptors: 1. GABA-A Contain chloride channels and contain different binding channel. It is an ionotropic receptor 2. GABA-B Receptors bound to G-proteins, therefore it is a metabotropic receptor 3. GABA-C It is also ionotropic with chloride channels, however it has a different subunit structure from other GABA A. A, Schematic diagram of the synthesis and transport of GABA (g-aminobutyric acid) at synapses. GABA is synthesized in inhibitory neurons from glutamate by the enzyme glutamic acid decarboxylase (GAD), and is transported into vesicles by a vesicular transporter (VGAT). GABA can be released either vesicularly or non-vesicularly (by reverse transport). GABA receptors are located at pre- and postsynaptic sites. GABAB receptors are metabotropic receptors that cause presynaptic inhibition by suppressing calcium influx and reducing transmitter release, and achieve postsynaptic inhibition by activating potassium currents that hyperpolarize the cell. Reuptake of GABA by surrounding neurons and glia occurs through the activity of GABA transporters (GAT). Subsequently, GABA is metabolized by a transamination reaction that is catalysed by GABA transaminase (GABA-T). GABA receptors differ in subunit composition and assembly. GABAA and GABAC receptors are closely related pentameric receptors that carry chloride; however, whereas GABAA receptors are composed of combinations of several subunit types, GABAC receptors are composed of only single or multiple -subunits. GABAB receptors are metabotropic receptors that exist as R1a, R1b and R2 isoforms, and are associated with G proteins. Native GABAB receptors are dimers composed of one R1 subunit and the R2 subunit. Composition of GABAA receptors Receptors are oligomeric assemblies of a large range of subunits, a1-6, b1-3, g1-3, d, e, p and q In vitro, the pharmacology of the natural receptors can be replicated with pentameric combinations of cloned receptor-subtypes, provided at least one of each of the , abg and d subunits is present, and it is currently thought that the dominant stoichiometry in vivo is 2abg. While the dominant a1b2g2 subtypes are present in both cerebellum and cortex, the a2bn g2 and a3bng2/3 assemblies are found mainly in the cortex and hippocampus, and the a5b3g2 and a5b3g3 are largely confined to the hippocampus. In the mammalian brain, the major subtype has the composition 2-a1,2-b2,1-g2. Glutamate Receptor Subtypes Subunits GluR 1GluA1-4 4 GluN1 GluR 5-7, NR1, GluK1-3 GluN2A-D KA1,2 NR2A-2D GluK4-5 GluN3A-B mGlu1 mGlu5 mGlu2 mGlu3 mGlu4 mGlu6-8 Metabotropic receptors of glutamate Ionotropic receptors of glutamate Ionotropic glutamate receptor subunit Ionotropic receptors of glutamate NMDA receptor as a coincidence detector : requirement for membrane depolarization 15 Gaba and glutamate therapy Physiological processes : Memory Learning Orientation Pathological processes : Epilepsy Ischemia Alzheimer Tranche d`Hippocampe CA1 CA3 Vm HI LUS Stimulation Gaba and Glutamate are both biosynthesized and broken in patways linked to the citric acid cycle (Krebs cycle). Glutamine (from glia) COOH COOH glutamic acid decarboxylase H2N GAD COOH glutammate H2N GABA transaminases GABA-T a-ketoglutarate citric acid cycle Succinic semialdehyde Succinic acid The metabolism of the principal CNS excitatory and inhibitory neurotrasmitters is intimately linked. Biosynthetic Pathway and Metabolism of GABA An example of an anticonvulsant agent working by preventing this feedback inhibition (succinic acid) is sodium valproate (Divalproex Sodium) Agonisti, parziali agonisti e antagonisti gabaergici che competono con il sito del GABA-A GABA AGONISTI OH Muscimolo H2N H2N O N 5-Aminomethyl-isoxazol-3-ol C OOH 4-Amino-butyric acid Dicentra cucullaria GABA-A ANTAGONISTI di origine naturale O O H O N H O O O Bicucullina 6-(6-Methyl-5,6,7,8-tetrahydro-[1,3]dioxolo[4,5-g ]-isoquinolin-5-yl)-6H-furo[3',4':3,4]-benzo[1,2-d][1,3]dioxol-8-one ANTAGONISTI di sintesi OCH3 N N NH O OH 4-[6-Imino-3-(4-methoxy-phenyl)-6H-pyridazin-1-yl]-butyric acid Ligands of GABA-A receptor, as allosteric modulators In addition to the GABA binding site, allosteric modulatory sites for numerous ligands have been identified on GABAA receptors ( benzodiazepines (BZs), barbiturates, ethanol, steroids, avermectins, picrotoxin, zinc cations, and loreclezole ), of which the BZ site has historically been of the most pharmacological interest. Agonists (positive allosteric modulators) give an increased ion flux, resulting in a greater inhibitory effect on the postsynaptic neurone, Inverse agonists (negative allosteric modulators) cause a decrease in the ion flux in response to GABA, Antagonists (neutral allosteric modulators), have no effect on the ion channel response to GABA but will competitively displace other BZ site ligands from the receptor. Ligands of GABA-A receptor Therapeutic effetcs of gabaergic modulation Agonists of the GABA allosteric site show anxiolytic, sleep inducer and muscle relaxed effects. Inverse agonists are anxiogenic. Antagonists have no effect on anxiety, sleep disorders or muscle contraction disorder. Classical BZ drugs for the treatment of anxiety and sleep inducer, such as Diazepam, are full agonists at the BZ site of the GABA-A receptor with no subtype selectivity. Although clinically useful, such compounds often exhibit undesirable effects, particularly sedation, ataxia, and potentiation with alcohol. There is also a risk of tolerance and dependence developing with chronic use. Since, neural inhibition is the principle role of GABA, therefore substances that block GABA can provoke seizures. Allosteric modulators (agonists) of the GABA-A receptor Barbiturates phenobarbital O HN O HN NH O O O pentobarbital NH O 5-Ethyl-5-(1-methyl-butyl)-pyrimidine-2,4,6-trione 5-Ethyl-5-phenyl-pyrimidine-2,4,6-trione S HN O NH O Thiopental, more as sodium salt 5-Ethyl-5-(1-methyl-butyl)-2-thioxo-dihydro-pyrimidine-4,6-dione Butalbital, 5-allyl-5-isobutylbarbituric acid, is a barbiturate with an intermediate duration of action. Butalbital is often combined with other medications, such as paracetamol (acetaminophen) or aspirin, and is commonly prescribed for the treatment of pain and headache. The various formulations combined with codeine are FDA approved for the treatment of tension headaches. Barbiturate activity Barbiturates depress the central nervous system, they have a wider and more powerful effect on the central nervous system than the other sedatives. The barbiturates can produce varying degrees of depression of the CNS, ranging from mild sedation to general anesthesia. In low doses barbiturates have a calming effect, and some of the barbiturates (e.g., phenobarbital) have demonstrated selective anticonvulsant properties. In moderate doses they produce a drunken euphoric state, similar to alcohol. Sedation and sleep result from increased doses, and even higher doses produce surgical anesthesia. Because of their ability to produce sedation and decrease sleep latency, barbiturates were popular in the treatment of insomnia prior to the advent of benzodiazepines. However, because of the high incidence of tolerance and physical dependence following chronic use and the relatively high danger of overdose, these drugs are rarely used today for the treatment of anxiety or sleep disturbances. Allosteric modulators (agonists) of the GABA-A receptor R O N H O Glutetimide R = H, 3-Ethyl-3-phenyl-piperidine-2,6-dione R = NH2, Amino-glutetimide O N O N H O O Talidomide Allosteric modulators (agonists) of the GABA-A receptor Benzodiazepines R N N N N Metaqualone X O N O H N CH3 N Cl N O Clordiazepossido (7-Chloro-5-phenyl-3H-benzo[e][1,4]diazepin-2-yl)-methyl-amine 4-ossido R1 Modificazioni chimiche al Clordiazepossido H N CH3 N a Cl H N O N Cl O N O H+/H2O b a) La funzione imino-metil-aminica non è indispensabile per l’attività b) L’ N->O in posizione 4 non è indispensabile per l’ attività PCl3 H N Cl 7-Chloro-5-phenyl-1,3-dihydro-2H-benzo[e][1,4]diazepin-2-one O N Relazione struttura-attività del nucleo benzodiazepinico • L’ azoto in posizione 1 puo’ essere alchilato con gruppi di moderato ingombro sterico o con funzioni salificabili per migliorare la solubilità in solventi acquosi. Le posizioni 1,2 possono essere impiegate per la formazione di una ulteriore struttura ciclica. • Il carbonile della posizione 2 non è indispensabile per l’ attività, il parziale carattere di doppio legame dell’ amide rende il ciclo a 7 termini quasi planare, mentre i due aromatici si trovano su due piani che formano un angolo compreso tra 58-86°. H N Cl Derivato non ossigenato O N Relazione struttura-attività del nucleo benzodiazepinico • La posizione 3 puo’ essere sostituita con gruppi polari e idrofilici, OH, COOH, i 3-idrossi derivati sono dei metaboliti attivi. • La maggior parte delle benzodiazepine presenta il doppio legame in posizione 4/5, queste posizioni sono state utilizzate per la formazione di un ulteriore struttura ciclica; il sostituente in 5 è un aromatico, generalmente fenilico indispensabile per l’attività, la ulteriore sostituzione in 2’ con alogeni ne incrementa l’attività. • L’aromatico della nucleo benzo-diazepincio è sempre sostituito in posizione 7 con gruppi elettronattattori, Cl, NO2, Br e puo’ essere sostituito con isosteri (tiofene). • Il nucleo diazepinico puo’ essere strutturato in modo diverso ma la disposizione dei due anelli aromatici deve rientrare nei termini indicati precedentemente. H O N Cl N 3 2’ Alcune benzodiazepine di uso terapeutico Ansiolitici, ipnoinducenti, antiepilettici e miorilassanti O N Cl N Diazepam 7-Chloro-1-methyl-5-phenyl-1,3-dihydro-2H-benzo[e][1,4]diazepin-2-one H N O2N O N Nitrazepam Flurazepam 7-Nitrol-5-phenyl-1,3-dihydro-2H-benzo[e][1,4]diazepin-2-one 7-Nitro-5-(2-fluoro-phenyl)-1-methyl-1,3-dihydro-2H-benzo[e][1,4]diazepin-2-one Flunitrazepam About the medical use of benzodiazepines Diazepam possesses anxiolytic, anticonvulsant, sedative, skeletal muscle relaxant and amnestic properties. It is commonly used for treating anxiety, insomnia, seizures, alcohol withdrawal, and muscle spasms. It may also be used before certain medical procedures (such as endoscopies) to reduce tension and anxiety, and in some surgical procedures to induce amnesia. Diazepam has a half-life of 20–50 hours, and desmethyldiazepam has a half-life of 30–200 hours and is considered to be a long acting benzodiazepine. Diazepam is metabolised via oxidative pathways in the liver via the cytochrome P450 enzyme system. It has a biphasic half-life of 1–2 and 2–5 days, and has several pharmacologically active metabolites. The main active metabolite of diazepam is desmethyldiazepam (also known as nordazepam or nordiazepam). Diazepam's other active metabolites include temazepam and oxazepam. These metabolites are conjugated with glucuronide, and are excreted primarily in the urine. The United States Pharmacopoeia lists diazepam as soluble 1 in 16 of ethyl alcohol, 1 in 2 of chloroform, 1 in 39 of ether, and practically insoluble in water. Nitrazepam is a hypnotic drug with sedative and motor impairing properties, anxiolytic, amnestic, anticonvulsant, and skeletal muscle relaxant properties. Nitrazepam is long acting and is sometimes used in patients who have difficulty in maintaining sleep. Flunitrazepam is considered to be one of the most potent benzodiazepine hypnotic on effect, rather than on dose basis; i.e., its hypnotic effect is considered to be one of the most strongly pronounced of all benzodiazepine hypnotics available. Abuse of flunitrazepam among drug addicts is considerable and any possession of flunitrazepam without a valid prescription is illegal. Alcune benzodiazepine di uso terapeutico Ansiolitici, ipnoinducenti, antiepilettici e mirilassanti H N O OH Cl N Cl Lorazepam 7-Chloro-5-(2-chloro-phenyl)-3-hydroxy-1,3-dihydro-2H-benzo[e][1,4]diazepin-2-one Oxazepam Lorazepam is a benzodiazepine drug with short to medium duration of action. It has all five intrinsic benzodiazepine effects: anxiolytic, sedative/hypnotic, anticonvulsant and muscle relaxant, to different extents. It is a powerful anxiolytic. It is a unique benzodiazepine insofar as it has also found use as an adjunct antiemetic in chemotherapy. Pure lorazepam is an almost white powder that is nearly insoluble in water and oil. In medicinal form, lorazepam is mainly available as tablets and a solution for injection but in some locations it is also available as a skin patch, an oral solution and a sublingual tablet. Lorazepam injectable solution is administered either by deep intramuscular injection or by intravenous injection. The injectable solution comes in 1 mL ampoules containing 2 mg or 4 mg lorazepam. The solvents used are polyethylene glycol 400 and propylene glycol. As a preservative, the injectable solution contains benzyl alcohol. Benzodiazepine di uso terapeutico Ansiolitici, ipnoinducenti, antiepilettici e mirilassanti N N N Cl N Alprazolam 8-Cloro-6-fenil-1-metil-4H-1,2,4-triazolo-4,3a-1,4-benzo [e]diazepina •Triazolam, 6(2-chloro-phenyl)-Alprazolam •Adinazolam, (1-dimethy-amino-methyl)-Alprazolam N N Cl N F Midazolam 8-Cloro-6-(2-fluorofenil)-1-metil-4H-imidazo-1,5-a-1,4-benzo-[e]diazepina Alprazolam, also known under the trade names Xanax and Niravam, is a short-acting drug of the benzodiazepine class. The first indication for which alprazolam was approved was panic disorder. It is generally used to treat moderate to severe anxiety disorders, panic attacks, and as an adjunctive treatment for anxiety associated with clinical depression. Alprazolam is FDA-approved for the short term treatment (up to 8 weeks) of panic disorder, with or without agoraphobia. Alprazolam is very effective in preventing moderate to severe anxiety, essential tremor, panic attacks and other types of convulsive behaviors. Triazolam (marketed under brand names Halcion, Novodorm, Songar) is a benzodiazepine possessing pharmacological properties similar to that of other benzodiazepines, but it is generally only used as a sedative to treat insomnia. Insomnia can best be described as a difficulty falling asleep, frequent awakening, early awakenings or a combination of each. Triazolam is a short acting benzodiazepine and is sometimes used in patients who have difficulty in falling asleep. Short half life hypnotics such as triazolam are not effective in patients who suffer from frequent awakenings or early wakening due to their very short half life. Triazolam has a very high risk of dependency with chronic users often taking exceedingly high daily doses. Midazolam (marketed under brand names Dormicum, Flormidal) is a drug which is a benzodiazepine derivative. It has powerful anxiolytic, amnestic, hypnotic, anticonvulsant, skeletal muscle relaxant and sedative properties. It is considered a short-acting benzodiazepine, with a short elimination half-life. It is therefore a very useful drug to use for short minor procedures such as dental extraction. Because of midazolam's extremely short duration, midazolam is not used for patients who have trouble staying asleep through the night; moderate to long acting benzodiazepines like temazepam, nitrazepam, flunitrazepam and lormetazepam are used for those purposes. Midazolam is also indicated for the acute management of aggressive or delirious patients and also is sometimes used for the acute management of seizures such as status epilepticus. Long term use for the management of epilepsy is not recommended however, due to the significant risk of tolerance which renders midazolam and other benzodiazepines ineffective and as well the significant side effect of sedation. Midazolam and other benzodiazepines cause a deterioration in sleep quality. Cyproheptadine may be superior to nitrazepam in the treatment of insomnia as it enhances sleep quality based on EEG (Electroencephalography) studies. Cyproheptadine (usually as cyproheptadine hydrochloride, trade name Periactin) is an antihistaminic and antiserotonergic agent. It acts as a 5-HT2 receptor antagonist and also blocks calcium channels. Summary of benzodiazepines activity and side effects •The benzodiazepines are currently the most important class of drugs for treatment of anxiety and sleep disorders. •Benzodiazepines share the sedative-hypnotic properties, but produce fewer side effects than barbiturates. •Like barbiturates, benzodiazepines have also been reported to produce anticonvulsant effects. •In addition, these drugs are used clinically as muscle relaxants, antiepilieptic agents, and to produce sedation before operative procedures. •The antianxiety effects of benzodiazepines are more selective than those of other sedative-hypnotics, they relieve anxiety at lower doses and thus produce minimal sedation and motor impairment. Side effects of benzodiazepins consider: tolerance and physical dependence, •amnesia and oversedation such as potentiation of alchool and other drugs. Ó) Inverse agonist of the GABA-A receptor, negative modulator O O O N N H O 4-Ethyl-6,7-dimethoxy-9H-b-carboline-3-carboxylic acid ethyl ester O N ◊ Antagonist of the GABA-A receptor Cl N N N O O Diazepam N F O Flumazenil 8-Fluoro-5-metil-5,6-diidro-4H-imidazo-1,5-a-1,4-benzo-[e]-diazepin-6-one-3-carbossilico acido etil estere Atypical benzodiazepine receptor ligands •While unselective full agonist BZs such as diazepam are anxiolytics with sedative side effects apparent at higher doses, the BZ site full agonist zolpidem that has some binding selectivity for a1-containing receptors over other GABA-A subtypes is a clinical hypnotic, implicating a1-containing subtypes in the sedative response. O CH3 N N N CH3 H3C N O N CN N N Zalepon N-[3-(3-Cyano-pyrazolo[1,5-a]-pyrimidin-7-yl)-phenyl]-N-ethyl-acetamide H3C Zolpidem N,N-Dimethyl-2-(6-methyl-2-p-tolyl-imidazo[1,2-a]-pyridin-3-yl)-acetamide Recently it is confirmed that Zolpidem and Zalepon are agonists “with high affinity for a1 subunit-containing GABAA receptors, it has very high intrinsic activity and is a hypnotic that does not induce physical dependence”. Other ligands of the GABA-A receptor Ramelteon In 2005, the FDA approved the melatonin receptor antagonist RAMELTEON for the treatment of insonnia, rappresenting the first novel mechanism to be approved in 35 years and the only hypnotic not classified as a controlled substance. Ramelteon, marketed as Rozerem by Takeda Pharmaceuticals North America, is the first in a new class of sleep agents that selectively binds to the MT1 and MT2 receptors in the suprachiasmatic nucleus (SCN), instead of binding to GABA A receptors, such as with drugs like zolpidem, eszopiclone, and zaleplon (S)-N-[2-(1,6,7,8-tetrahydro-2H-indeno-[5,4-b]furan-8yl)ethyl]propionamide Melatonin Ramelteon is a melatonin receptor agonist with both high affinity for melatonin MT1 and MT2 receptors and selectivity over the MT3 receptor. Ramelteon demonstrates full agonist activity in vitro in cells expressing human MT1 or MT2 receptors, and high selectivity for human MT1 and MT2 receptors compared to the MT3 receptor. The activity of ramelteon at the MT1 and MT2 receptors is believed to contribute to its sleep-promoting properties, as these receptors, acted upon by endogenous melatonin, are thought to be involved in the maintenance of the circadian rhythm underlying the normal sleep-wake cycle. Ramelteon has no appreciable affinity for the GABA receptor complex or for receptors that bind neuropeptides, cytokines, serotonin, dopamine, noradrenaline, acetylcholine, and opiates. The metabotropic GABA-B receptors constitute a distinct subclass of receptors for the inhibitory amino acid GABA (Bowery, 1993). GABA-B receptors have been shown to interact with G proteins of the Gi/Go family and to activate different signalling mechanisms, including stimulation of K+ conductance, inhibition of Ca2+ channel activities and modulation of cyclic AMP formation. It should be pointed out that the exact location and the functional role of the GABA-B receptor are still unknown. However, it appears to be located on the pre- and postsynaptic which regulate GABA release. gabaergic neurons The metabotropic GABAB receptor • These receptors are GPCRS • Largely presynaptic, inhibit transmitter release • Most important role is in the spinal cord • Baclofen, an agonist at this receptor, is a muscle relaxant 46 Metabotropic GABA-B receptors (-) Baclofen was an agonist of the GABA-B receptor (+) Baclofen is less active It is believed that Baclofen, acting like GABA, blocks the activity of nerves within the part of the brain that controls the contraction and relaxation of skeletal muscle. Baclofen Phaclofen OH H2N [3-Amino-2-(4-chloro-phenyl)-propyl]-phosphonic acid O Cl (R) 4-Amino-3-(4-chloro-phenyl)-butyric acid H2N OH O P OH (3-Amino-propyl)-phosphonic acid OH O P OH H2N Cl GLUTAMATE therapy Memantine – Innovation for Alzheimer Patients It is the only drug of the NMDA glutamate receptor Amantadine, an atypical antiparkinson drug 2. Patological condition of lerning 1. Physiological condition of lerning 3. NMDA protection with the drug MEMANTINA Fine presentazione