Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
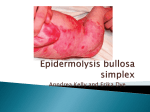
Epidermolysis bullosa. Part 1: causes, presentation and complications Elizabeth Pillay Abstract This article is the first in a series of three focusing on the causes, clinical presentation, complications and care of adult patients affected by epidermolysis bullosa (EB), a group of rare genetic skin fragility disorders. Although the condition is rare, in some cases it presents extreme challenges both to those affected and those involved in the care of the EB patient; therefore, these articles may have relevance for other long-term disorders. While there is a wealth of information regarding the ‘science’ of EB there is dearth of information regarding the care of the adult EB patient, and this series of articles will endeavour to fill that gap. This article focuses mainly on those patients affected with the most severe form of EB found in the adult group, recessive dystrophic epidermolysis bullosa; with the part two looking at the care of the adult with EB from the nursing perspective, including wound management, and the experiences of a specialist EB psychotherapist being presented in the final article of the series. Readers will thus have an opportunity to gain an overall view of this difficult condition. Key words: DebRA disorders n Dermatology n Epidermolysis bullosa E pidermolysis bullosa (EB) is an umbrella term for a group of rare genetic skin blistering disorders characterized by blister formation in response to minimal trauma or friction. Fragility can also extend to some of the internal mucosa and the eye in some forms of EB (Uitto et al, 2000). The effects of EB range from death in early infancy in the most severe forms, to other patients experiencing a life of increasing disability. In the ‘milder’ forms a normal life span is anticipated, although some disability and pain is experienced by most patients (Fine et al, 2004a). In severe forms multiple squamous cell carcinomas are a common cause of early death. All forms of EB result from mutations in genes, which encode for the formation of structural proteins at the dermal-epidermal junction/basement membrane zone. The weakened, or in some cases absent structures lead to ease of separation of the layers of skin and consequent fragility (Uitto and Pulkkinen, Elizabeth Pillay is EB Nurse Consultant and Team Leader for DebRA Adult Nursing Team, DebRA UK and Guys and St Thomas’ NHS Foundation Trust, London Accepted for publication: February 2008 292 n Genetic n Skin mutations causing recessive dystrophic EB (RDEB), apart from their rarity, are many and varied, and identifying the causative mutation is a complex, highly-skilled procedure carried out by a few specialist centres globally. The incidence of EB has not been fully recorded, although there are thought to be approximately 5000 individuals in the UK suffering from all forms of EB (DebRA, 2008). The EB registry in the United States reports cases of EB of all forms is 54 per million live births (Marinkovitch and Pham, 2006). Atherton and Denyer (2003) give a prevalence in the UK of 17–32 per million for all forms of EB simplex and estimate there to be 20 per million infants born with junctional EB. Horn et al (1995) give a Scottish prevalence of 21.4 per million for all forms of dystrophic EB. Forms of EB 2001). The type and severity of EB depend on which gene is affected (Uitto et al, 2000), and the variety in clinical presentation is caused by the many different causative mutations. EB is a genetic condition and can be inherited either recessively or dominantly (Priestly et al, 1990). The more severe forms are generally recessively inherited. In a dominantly inherited disorder an affected parent will have passed the faulty gene to their child. There is a 1 in 2 chance with each pregnancy that a child will inherit the affected gene, which overwhelms the normal gene inherited from the unaffected parent.There is no ‘carrier’ status in a dominant condition. Not infrequently a new mutation known as a ‘de-novo’ mutation occurs. In this situation the patient will have no family history of the condition, but will have developed the mutation spontaneously (Hashimoto et al, 1999; Atherton and Denyer, 2003). In a recessively inherited disorder each of the parents must carry the affected gene.With each pregnancy the carrier parents have together there is a 1 in 4 chance of an affected child being conceived (McLean, 2000); thus, a person with EB may have unaffected siblings. Unlike some other recessive conditions (e.g. sickle cell anaemia), carrier testing cannot be offered to the general antenatal population as the genetic There are a number of differing forms of the condition classified by the level at which skin cleavage occurs.There are three major groupings of EB: simplex, junctional and dystrophic. EB Simplex (EBS) In EBS the weakened structures are within the epidermis itself.This is the most common form of EB, and although the fragility extends all over the skin, blistering is most troublesome in the areas exposed to most friction, i.e. the hands and feet (Figures 1 and 2). Blistering is exacerbated by heat and healing is without scarring. EBS is overwhelmingly dominantly inherited. EBS is almost always compatible with a normal life-span (Atherton and Denyer, 2003; Pillay and Graham-King, 2007). Junctional EB (JEB) There are two major forms of JEB, with Herlitz JEB commonly leading to death in early infancy, while non-Herlitz JEB is usually compatible with a near normal life-span (Atherton and Denyer, 2003). In non-Herlitz JEB the most common problems are areas of chronic ulceration (Figure 3), nail dystrophy and loss, corneal erosions and scarring, alopecia, and dental problems due to improperly formed dental enamel (Pai and Marinkovich, 2002). British Journal of Nursing, 2008, Vol 17, No 5 dermatology nursing Fragility of the genitourinary tract is commonly found in JEB and can lead to such problems as urethral stenosis (Fine et al, 2004b). The weakened structures are to be found at the level of the lamina lucida of the basement membrane zone. JEB is always recessively inherited (Atherton and Denyer, 2003). Figure 2. Blistering caused by epidermolysis bullosa simplex Weber-Cockayne (Marinkovitch, 2007). Figure 3. Chronic ulceration typically seen in junctional epidermolysis bullosa. Tidman, 2002). The cornea and conjunctiva of the eye are also affected (Tong et al, 1999). In severe RDEB the most common cause of death is as a result of multiple aggressive squamous cell carcinomas (Uitto et al, 2002). Healing with scarring is a prominent feature of this form of EB, and can lead to marked disability. RDEB is a very variable disorder, and the clinical picture is determined by the causative genetic mutation and the amount of collagen Vll the individual is able to make (Atherton and Denyer, 2003). Figure 4. Typical blistering found in dystrophic epidermolysis bullosa. Dystrophic EB (DEB) DEB can be recessively or dominantly inherited with the most severe effects generally occurring when the condition is recessively inherited (Horn and Tidman, 2002). In RDEB patients frequently have blistering and wounds over a large proportion of their body surface with the fragility extending to some of the internal mucosa and the eye. In dominant dystrophic epidermolysis bullosa skin and mucosal effects are generally less severe and many patients are able to lead a near normal life (Fine et al, 2000; Pai and Marinkovich, 2002). Recessive dystrophic EB (RDEB) The affected gene in all forms of DEB is the one that encodes for the protein collagen Vll. Collagen Vll is a crucial component of the anchoring fibrils which act like ‘velcro’ in binding the epidermis to the dermis. If the fibrils are absent or diminished this leads to separation of the epidermis from the dermis in response to minimal trauma or friction (Figure 5) (Wojnarowska, 1998). Thus, blisters are formed with ease. These blisters go on to form wounds, some healing within days, with other blister sites forming chronic wounds. This fragility also affects some of the internal mucosa, most notably of the mouth, oropharynx, oesophagus and the anal margins (Horn and Symptoms, complications and management RDEB The skin Patients with severe RDEB will present with extensive skin involvement. They will have blisters and wounds in various stages of healing. These wounds can occur anywhere on the body, but tend to occur on areas subject to repeat trauma e.g. hands, feet and bony prominences (Wojnarowska, 1998). Wound healing is compromised by some of the other effects of RDEB as outlined below. Figure 1. Epidermolysis bullosa simplex Dowling Meara (Marinkovitch, 2007). British Journal of Nursing, 2008, Vol 17, No 5 Wound management will be covered more fully in the second article of this series; however, wound care in RDEB has two major objectives: the provision of the optimal healing environment for wounded areas, and protection against further trauma. All dressings used must be non-adherent both to the wound bed and the fragile peri-wound skin. Commonly used primary dressings are the soft-silicone range; Mepitel® and the various forms of Mepilex® (Mölnlycke Health Care Ld, Dunstable) and dressings coated with lipido-colloid, Urgotul® and Urgotul® SSD (Urgo Ltd, Loughborough). Dressing changes for a severely affected EB patient can take many hours. All blisters must be lanced and drained to prevent extension through the improperly bound skin layers. Chronic wounds occur in many patients (Figures 6 and 7), most notably in areas where repeated wounding is a feature and very fragile areas may heal and break down again with rapidity (Figures 8 and 9). The ability of these patients to heal is compromised by complications such as malnutrition and anaemia (Mellerio et al, 2007). Local factors compromising wound healing are increased bio-burden and wound infections as the protective functions of the skin are lost (Sibbald et al, 2005). Over-granulation is common, as is the presence of necrotic material, commonly slough. Pruritus can be an 293 Figure 5. Absence or diminishing of anchoring fibrils, leading to separation of the epidermis from the dermis in response to minimal trauma or friction in recessive dystrophic epidermolysis bullosa (EB). This is due to reduced production of protein collagen VII, which is encoded by the gene affected in all types of dystrophic EB. Epidermis Basement membrane Dermis Separation of epidermis and dermis (blister formation) Epidermal cells Basement membrane Collagen Vll anchoring the basement membrane to dermal structures Dermis overwhelming problem in RDEB and is the cause of many new blisters and the breakdown of almost healed wounds. Treatment includes application of bland emollients to dry skin – endeavouring to keep the patient cool – and oral anti-pruritics (Schober-Flores, 2003; Mellerio et al, 2007). potential to incur damage while undergoing procedures; this can be both cutaneous and that occasioned by anaesthetic instruments and procedures (Griffin and Mayou, 1993; Iohom and Lyons, 2001). Gastrointestinal complications Wounds heal with atrophic scarring leading to contractures, most notably of the hands and feet. Scarring and contractures lead to the loss of functional digits as both hands and feet are ‘cocooned’ in scar tissue – this is known as a mitten deformity or psuedosyndactyly (Figure 10). Surgical release of contractures of the hand is undertaken, although contractures return, and repeat surgery is required after a variable time (Figure 11) (Fine et al, 2005). Peri-operative and anaesthetic management of these patients is a challenge because of the Gastrointestinal complications begin in the mouth, where, although there are no primary problems with enamel, such as those seen in JEB, microstomia – as a result of scarring and the fragility of the oral mucosa – lead to poor dental hygiene with resultant loss of dentition (Mason, 2003). Microstomia means that dental access is poor and dental treatment may have to be carried out by a specialist practitioner under general anaesthetic. Loss of teeth makes mastication difficult and dentures are contraindicated because of potential damage to fragile gums, although more recently dental implants have been shown Figure 6. Chronic wound to the back in recessive dystrophic epidermolysis bullosa. Figure 7. Chronic wounds of many years duration in recessive dystrophic epidermolysis bullosa. Contractures and scarring 294 to be a solution in some cases (Penarroch et al, 2007). Malnutrition is common and is compounded by increased nutritional requirements as a result of multiple open wounds and inflammation; the wounds bleed and exudate loss leads to a loss of valuable nutrients (Mellerio et al, 2007). Poor intake as a result of blistering and pain in the mouth, and blistering and narrowing of the oesophagus, further exacerbates the problem, with chronic constipation being a big disincentive to eating. All the National Care Group centres now have an EB specialist dietician and dentist. A soft or liquidized diet may be necessary with supplementary feeding being common. Many younger patients now have a gastrostomy placed to allow for ease of supplementary feeding and medication (Hayne et al, 1996). Dysphagia following narrowing of the oesophagus is treated by balloon dilatation of oesophagus under X-ray, and may need to be repeated regularly (Azizkhan, 2006). Gastric reflux is a common problem exacerbating oesophageal damage and requires preventative management with proton-pump inhibitors (Azizkhan et al, 2006). Chronic constipation occurs as a result of blistering of the anal margins, causing pain on defecation. This can lead to the patient learning to withhold faeces as a child, which in turn causes poor bowel tone. Management includes increasing dietary fibre, avoidance of drugs known to cause constipation, such as codeine, and the giving of appropriate laxatives (Haynes et al, 1996; Mellerio et al, 2007). Additionally, in some rare cases, the gut can show inflammatory changes leading to diarrhoea and other associated symptoms (Shah et al, 2007). Chronic anaemia Occurs as a result of blood loss from open wounds, poor nutritional intake of iron and ‘anaemia of chronic disease’ (Mellerio et al, 2007). Management includes encouraging the patient to increase their dietary iron intake while oral iron supplementation as a liquid may help, although poor absorption can mean it is unsuccessful in some cases. Some patients are reluctant to take iron as it may exacerbate constipation. Intravenous Venofer® (SynerMed, Surrey) – a form of iron sucrose – has been very successful in the management of anaemia in EB although peripheral venous access is often difficult due to scarring (Atherton et al, 1999; Mellerio et al, 2007). Dilated cardiomyopathy Dilated cardiomyopathy has been found in a small number of children with severe RDEB British Journal of Nursing, 2008, Vol 17, No 5 dermatology nursing Figure 8. Chronic wounds in severe recessive dystrophic epidermolysis bullosa. Figure 9. Blistering in severe recessive dystrophic epidermolysis bullosa. appear little different from other wounds the patient may have. Additionally, the tumours have no common appearance and identification depends upon the patient, their care team and on the expertise of the examining EB clinician who will be familiar with the varied presentations of these tumours. The biopsy specimen requires a specialist pathologist who is able to differentiate a malignancy from the skin pathology of EB.Treatment is surgical with wide local excision with grafting. Adjunctive therapies may include the use of radiotherapy and rarely chemotherapy (Mallipeddi, 2002). and can lead to early mortality (Sidwell et al, 2000).The aetiology is not properly understood but possible causes are deficiencies in carnitine and selenium, infection, chronic anaemia and/ or iron overload, occurring as part of the disease process of severe RDEB. Management includes regular monitoring with echocardiogram to detect cardiomyopathy,monitoring of nutritional status with appropriate supplementation with prompt treatment of anaemia or iron overload (Sidwell et al, 2000). secondary to the chronic inflammation and antigen stimulation seen in severe EB (Mann et al, 1988; Gündüz et al, 2000) n Immunoglobulin A (IgA) nephropathy developing as a result of deposits of the protein IgA in the glomeruli of the kidney (Mellerio, 2007). Obstructive uropathy commonly arises at the vesicoureteric junction and in the urethra, and can affect the renal function (Mann et al, 1988; Gündüz et al, 2000; Fine et al, 2004b). Some patients will develop eroded areas in the groin/vulval areas.These are particularly difficult to heal because of the problems of dressing retention and constant chafing from walking and underwear. This, along with failure of puberty, may affect the ability of the patient to have a sexual relationship, although some severely affected EB patients have become parents (Mallipeddi et al, 2003; Lucky et al, 2007). Pain Osteoporosis/osteopenia Occurs in most patients as a result of a lack of weight-bearing exercise, and generalized inflammation which probably increases boneturnover (Mellerio et al, 2007). Management includes increasing weight-bearing exercise where possible, avoidance of long-term steroid use, and the giving of vitamin D and calcium supplements and bisphosphonates. Dual energy X-ray absorptiometry scans and plain radiographs should be carried out annually (Keane et al, 2001; Fewtrell et al, 2006). Corneal abrasions and ulceration Occur in some patients as a result of the fragility of the conjunctiva and cornea. These can be extremely painful and are managed by nursing the patient in a darkened room with a soft patch over the affected eye. Substantial analgesia may be required. Prevention is the application of lubrication and artificial tears to the eye (Tong et al, 1999). Genitourinary tract Both RDEB and JEB can affect the genitourinary tract in a number of ways, following fragility, scarring and infection in this area. Conditions leading to acute or chronic renal failure in EB are: n Glomerulonephritis secondary to streptococci or other infection (Mann et al, 1988) n Renal amyloidosis, which is a build-up of abnormal protein deposits in the kidney British Journal of Nursing, 2008, Vol 17, No 5 Pain in EB is multi-factorial and may be as a result of chronic wounds, contractures, osteoporosis, dental pain, pain from dysphagia and blistering of the oesophagus, pain from corneal ulcerations, pain on defecation and procedural pain, e.g. at dressing changes. Careful assessment of the type and cause of pain is needed with referral to pain teams as required (Herod et al, 2002; Mellerio et al, 2007). Psychological issues Psychological issues with depression and other emotional issues are common in EB, with frustration and non-concordance being a common feature in many EB patients. Given the severity of the disorder, with its unremitting ability to inflict pain, suffering and disability, this is not surprising (Lucky et al, 2007). The added dimension of the inevitability (for Squamous cell carcinoma Squamous cell carcinoma is a later – and in the severe forms, an inevitable – complication of RDEB, with multiple primaries occurring most notably on the bony prominences. The cause of this is not clearly understood and research is ongoing (Uitto et al, 2000). This leads to a shortened life span for severely affected individuals with tumours occurring as early as the teenage years, although this is rare (Ayman, 2002), with the majority of first tumours occurring in the third or fourth decades (Fine et al, 1999). Average prognosis is 5 years from diagnosis of first tumour to death, despite all treatment, and 55% of patients with severe RDEB will die of metastatic SCC by age 40 (Fine et al, 1999); however, this is a retrospective view and there are wide individual variations. Constant vigilance with regular skin surveillance in the multidisciplinary EB clinic, and early biopsy of suspect areas is routine management. Tumours can be extremely difficult to identify in the EB patient, both because of the unusual appearance of EB skin, but also because some Figure 10. Mitten deformity or psuedosyndactyly. Figure 11. Hand surgery to release contractures. 295 severely affected patients) of the development of aggressive squamous cell carcinoma is a further contributor to emotional distress. Both psychological and pharmacological treatments are offered. This area will be fully explored in the third part of this series of articles. Services for people with EB Specialist centres housing extensive multidisciplinary teams headed by a dermatologist can be found at St Thomas’ and Heartlands Hospitals, Solihull (for adults), with paediatric services provided at Great Ormond Street, London, and Birmingham Children’s Hospitals. These services are funded by the National Care Group and as such there is no funding implication for local primary care trusts when referring patients. Both paediatric and adult services are provided at Edinburgh Royal Infirmary. A team of 11 specialist EB nurses covering both paediatrics and adults are funded by the charity DebRA. The role of these nurses is primarily the support of patients with EB and their families in the community. This means the team visits patients throughout the UK and support and advise local healthcare providers. A further five nurses are funded through National Care Group and carry out a role similar to the DebRA nurses. DebRA is the national charity that supports individual and families affected by EB, and was founded in 1978 by a group of parents of children born with EB. DebRA provides an expert team of nurses and social care workers, and both commissions and funds research projects into the causes, care and potential cures for EB worldwide (Hon, 2003). Conclusion This series aims both to inform the wider nursing community about EB, and also dispel the myth that ‘it’s only the skin’. ‘Inherited epidermolysis bullosa (EB) is one of the most devastating chronic diseases known to mankind’ (Fine et al, 2004b).The severity of the condition, with all its complexities and the possibility that routine care can be so damaging, provide special challenges for nurses involved in the care of such patients. At an emotional level nurses can be challenged by the extreme suffering that can be involved, and in the steady decline in patients they may have known for many years. All EB patients and their families benefit from, and should be seen in, specialized centres familiar with the many complications of EB, and appropriate management. Much can be done to improve the quality of life of people affected by EB, and although a cure is not yet possible, expert symptom management and 296 prevention of complications, leads both to increased life expectancy and improved quality of life, both for the individual and the family. People living with EB, whatever the degree of severity, do so in the community where local services are primary in meeting their needs, and care partnerships with the local primary healthcare teams and the specialized centres, are BJN vital to good service provision. Atherton DJ, Cox I, Hann I (1999) Intravenous iron (III) hydroxide-sucrose complex for anaemia in epidermolysis bullosa. Br J Dermatology 140(4):773 Atherton D, Denyer J (2003) Epidermolysis bullosa: an outline for professionals. DebRA, Berkshire Ayman T, Yerebakan O, Ciftcioglu MA, Alpsoy E (2002) A 13-year-old girl with recessive dystrophic epidermolysis bullosa presenting with squamous cell carcinoma. Pediatr Dermatol 19(5):436–8 Azizkhan RG, Stehr W, Cohen AP et al (2006) Esophageal strictures in children with recessive dystrophic epidermolysis bullosa: an 11-year experience with fluoroscopically guided balloon dilatation. J Pediatr Surg 41(1): 55–60 DebRA (2008) What is EB? DebRA, Berkshire. Available at: http://tinyurl.com/38v6at (last accessed 5 March 2008) Fewtrell MS, Allgrove J, Gordon I et al (2006) Bone mineralization in children with epidermolysis bullosa. Br J Dermatol 154(4): 959–62 Fine JD, Bauer A, McGuire J, Moshell A (1999) Cancer and inherited epidermolysis bullosa. In: Epidermolysis Bullosa. John Hopkins University Press, Baltimore, MA: 175–92 Fine JD, Eady RAJ, Bauer EA et al (2000) Revised classification system for inherited epidermolysis bullosa: Report of the Second International Consensus Meeting on diagnosis and classification of epidermolysis bullosa. J Am Acad Dermatol 42(6): 1051–66 Fine JD, Johnson LB, Weiner M, Suchindran C (2004a) Assessment of mobility, activities and pain in different sub-types of epidermolysis bullosa. Clin Exp Dermatol 29(2): 122–7 Fine JD, Johnson LB, Weiner M et al (2004b) Genitourinary complications of inherited epidermolysis bullosa: experience of the national epidermylosis bullosa registry and review of the literature. J Urol 172(5 Pt 1): 2040–4 Fine JD, Johnson LB,Weiner M (2005) Pseudosyndactyly and musculoskeletal contractures in inherited epidermolysis bullosa: experience of the National Epidermolysis Bullosa Registry, 1986-2002. J Hand Surg (Br) 30(1):14–22 Griffin RP, Mayou BJ (1993) The anaesthetic management of patients with dystrophic epidermolysis bullosa. A review of 44 patients over a 10 year period. Anaesthesia 48(9): 810–15 Gündüz K, Vatansever S, Türel A, Sen S (2000) Recessive dystrophic epidermolysis bullosa complicated with nephrotic syndrome due to secondary amyloidosis. Int J Dermatol 39(2): 151–3 Hashimoto I, Kon A, Tamai K, Uitto J (1999) Diagnostic dilemma of ‘sporadic’ cases of dystrophic epidermolysis bullosa: a new dominant or mitis recessive mutation? Exp Dermatol 8(2): 140–2 Haynes L, Atherton DJ, Ade-Ajayi N, Wheeler R, Kiely E (1996) Gastrostomy and growth in dystrophic epidermolysis bullosa. Br J Dermatol 134(5): 872–9 Herod J, Denyer J, Goldman A, Howard R (2002) Epidermolysis bullosa in children: pathophysiology, anaesthesia, and pain management. Paediatr Anaesth 12(5): 388–97 Hon J (2003) Debra and epidermolysis bullosa. The Leg Ulcer Forum Journal 17: 16–1 Horn H, Priestley G, Tidman M (1995) Epidemiology of epidermolysis bullosa in Scotland. Br J Dermatol 133: 1005 Horn H, Tidman M (2002) The clinical spectrum of dystrophic epidermolysis bullosa. Br J Dermatol 146(2): 267–74 Iohom G, Lyons B (2001) Anaesthesia for children with epidermolysis bullosa: a review of 20 years’ experience. Eur J Anaesthesiol 18(11): 745–54 Keane F, Fine J-D, Pillay E, Stein I, McGrath J, Eady R (2001) Osteopaenia and osteoporosis in recessive dystrophic epidermolysis bullosa. Br J Dermatol 145(59): 12–14 Lucky AW, Pfendner E, Pillay E, Paskel J, Weiner M, Palisson F (2007) Psychosocial aspects of epidermolysis bullosa: Proceedings of the IInd International Symposium on Epidermolysis Bullosa, Santiago, Chile, 2005. Int J Dermatol 46(8): 809–14 McClean I (2000) Genetics of EB. DebRA, Berkshire Mallipeddi R (2002) Epidermolysis bullosa and cancer. Clin Exp Dermatol 27(8): 616–23 Mallipeddi R, Pillay E, Bewley S et al (2003) Pregnancy in epidermolysis bullosa. Br J Dermatol 149(Suppl 64): 51 Mann JF, Zeier M, Zilow E et al (1988) The spectrum of renal involvement in epidermolysis bullosa dystrophica hereditaria: report of two cases. Am J Kidney Disease 11(5): 437–41 Marinkovitch M (2007) Epidermolysis Bullosa. emedicine. Available at: http://tinyurl.com/2hjlhb (last accessed 5 March 2008) Mason C (2003)The mouth and teeth, in clinical management of children and adults with epidermolysis bullosa. DebRA, Berkshire Mellerio J (2007) Personal Communication. Mellerio J, Weiner M, Denyer JE et al (2007) Medical management of epidermolysis bullosa: Proceedings of the IInd International Symposium on Epidermolysis Bullosa, Santiago, Chile, 2005. Int J Dermatol 46(8): 795–800 Pai S, Marinkovich MP (2002) Epidermolysis bullosa: new and emerging trends. Am J Clin Dermatol 3(6): 371–80 Penarrocha M, Rambla J, Balaguer, Serrano C, Silvestre J, Bagan J (2007) Complete fixed prosthesis over implants in patients with oral epidermolysis bullosa. J Oral Maxillofac Surg 65(7): 103–6 Pillay E, Graham-King P (2007) Epidermolysis bullosa simplex (adults). DebRA, Berkshire Priestly G, Tidman M, Weiss J, Eady R (eds) (1990) Epidermolysis Bullosa: A Comprehensive Review Of Classification Management and Laboratory Studies. DebRA, Berkshire: 1–2 Schober-Flores C (2003) Epidermolysis bullosa: the challenges of wound care. Dermatol Nurs 15(2): 135–40, 141–4 Shah N, Freeman E, Martinez A (2007) Histopathological features of gastrointestinal mucosal biopsy specimens in children with epidermolysis bullosa. J Clin Pathol 60(7): 843–4 Sibbald RG, Zuker R, Coutts P, Coelho S, Williamson D, Queen D (2005) Using a dermal skin substitute in the treatment of chronic wounds secondary to recessive dystrophic epidermolysis bullosa. Ostomy Wound Manage 51(11): 22–46 Sidwell RU,Yates R,Atherton (2000) Dilated cardiomyopathy in dystrophic epidermolysis bullosa. Arch Dis Child 83(1): 59–63 Tong L, Hodgkins PR, Denyer J et al (1999) The eye in epidermolysis bullosa. Br J Ophthalmol 83(3): 323–6 Uitto J, Eady R, Fine JD, Feder M, Dart J (2000) The DEBRA International Visionary/Consensus Meeting on Epidermolysis Bullosa: summary and recommendations (review). J Invest Dermatol 114(4): 734–7 Uitto J, Pulkkinen L (2001) Molecular genetics of hereditable blistering disorders. Arch Dermatol 137(11):1458–61 Wojnarowska F (1998) Bullous eruptions. In: Rook A, Wilkinson D, Ebling F, eds. Textbook of Dermatology. 6th edn: 1817–97 Key Points nEpidermolysis bullosa (EB) is an ‘umbrella term’ for a group of rare genetic skin blistering disorders. nEB is a variable disorder, but at its most severe it is a complex condition with many complications. nSpecialist services are available at National Care Group funded centres in a number of hospitals in the United Kingdom. nDebRA is the charity that supports both research into the genetics of EB with the aim of finding a cure and also specialist nursing and social work teams. British Journal of Nursing, 2008, Vol 17, No 5