Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
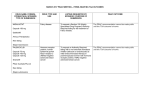
Fabry disease: clinical manifestations, diagnosis and therapy Second edition by Michael Beck Slides based on the book Beck M. Fabry disease: clinical manifestations, diagnosis and therapy. 2nd edition. Sponsored by Shire HGT The abbreviated PI can be found on slide 45 Overview of contents • The cause and discovery of Fabry disease • The genetic basis of Fabry disease • Signs and symptoms of Fabry disease • Enzyme trafficking and glycosylation • Clinical improvements following enzyme replacement therapy Defects in a single metabolic pathway lead to different lysosomal storage diseases Gal, galactose; GalNAc, N-acetyl-D-galactosamine; Glc, glucose; NeuAc, N-acetylneuraminic acid Reproduced from Beck & Ries, Fabry disease: clinical manifestations, diagnosis and therapy. Oxford: OCC Europe Ltd; 2001 with permission Globotriaosylceramide (Gb3) – the major lipid that accumulates in Fabry disease Gb3 is broken down into galactose and lactosylceramide by the enzyme α-galactosidase A Gal, galactose; Glc, glucose Reproduced from Beck & Ries, Fabry disease: clinical manifestations, diagnosis and therapy. Oxford: OCC Europe Ltd; 2001 with permission Structure of Gb3 Reproduced from Beck & Ries, Fabry disease: clinical manifestations, diagnosis and therapy. Oxford: OCC Europe Ltd; 2001 with permission Fabry disease was first described independently in 1898 by two physicians Johannes Fabry (1860–1930) William Anderson (1842–1900) Anderson, Br J Dermatol 1898;18:113–17; Fabry, Arch Derm Syph 1898;43:187–200 Angiokeratoma corporis diffusum in Johannes Fabry’s patient From Fabry, Arch Derm Syph 1916;123:294–307 Distribution of angiokeratoma corporis diffusum in William Anderson’s patient From Anderson, Br J Dermatol 1898;18:113–17 Inheritance of Fabry disease: the Lyon hypothesis Females inherit one X chromosome from each parent. In each cell, one of the X chromosomes is randomly inactivated, and the remaining X chromosome provides the genetic information. Reproduced from Beck & Ries, Fabry disease: clinical manifestations, diagnosis and therapy. Oxford: OCC Europe Ltd; 2001 with permission Location of the GLA gene that encodes the lysosomal enzyme α-galactosidase A Position of band Xq22 in the X chromosome Fabry disease is caused by a mutation in the GLA gene found in the Xq22 region of the X chromosome Exon structure of the GLA gene Adapted from Bishop et al., Proc Natl Acad Sci USA 1988;85:3903–7 with permission Signs and symptoms of Fabry disease • Fabry disease is a progressive multisystemic condition affecting both males and females • Many of the signs and symptoms begin during childhood or adolescence. In general, men are more severely affected than women, although some women may show severe manifestations of the disease • Typically, women develop signs and symptoms approximately 5–10 years later than men, although this is not invariably the case [1] • Patients with Fabry disease have an increased risk of death from renal, cardiovascular or cerebrovascular disease [2–3] 1. Mehta & Ginsberg, Acta Paediatr 2005;94 (Suppl 447);24–7 2. Colombi et al., Helv Med Acta 1967;34:67–83 3. MacDermot et al., J Med Genet 2001;38:750–60 4. MacDermot et al., J Med Genet 2001;38:769–75 Fabry disease has widespread effects throughout the body Proportion of patients with symptoms (%) Signs and symptoms of Fabry disease according to age at entry in FOS – the Fabry Outcome Survey Decade of life Reproduced from Mehta et al., Eur J Clin Invest 2004;34:236–42 with permission from Blackwell Publishing Neurological features of Fabry disease Peripheral nervous system • Peripheral neuropathy – Typically small fibre, painful – Impaired sweating – Temperature insensitivity Central nervous system • Cerebrovascular manifestations – Stroke – Transient ischaemic attacks • Epilepsy • Magnetic resonance imaging abnormalities – white matter lesions • Psychiatric/cognitive changes Sensory organs • Ear manifestations – Vertigo – Hearing loss – Tinnitus • Eye manifestations – Cornea verticillata – Subcapsular cataract – Tortuous vessels Reproduced from Mehta & Ginsberg, Acta Paediatr 2005;94 (Suppl 447):24–7 with permission from Blackwell Publishing Areas of the body affected by acroparaesthesiae in Fabry disease Pain begins in the hands and feet and radiates proximally to the shaded areas Magnetic resonance image showing the characteristic symmetrical high signal in the posterior thalamus in a patient with Fabry disease Reproduced from Ginsberg et al., Acta Paediatr 2006;95 (Suppl 451):57–62 with permission from Blackwell Publishing Lipid deposition in the kidneys begins in the glomerulus Lipid deposition in glomerular epithelium (blue arrows) and distal tubules (yellow arrow) with limited vascular endothelial involvement (orange arrow) Toluidine blue stain, original magnification x 100 Reproduced from Brady & Schiffmann, JAMA 2000;284:2771–5 © 2000 American Medical Association. All rights reserved Electron micrographs showing podocytes in the kidney in Fabry disease Glomerular podocytes appear enlarged and vacuolated Original magnification x 2200 Courtesy of Dr Jarl Ahlmén, Skövde, Sweden Lamellar myeloid bodies are evident in different regions of the glomerulus Original magnification x 10 500 Cardiac signs and symptoms and their cause in Fabry disease Symptom Leading cause Dyspnoea Diastolic LV dysfunction Pulmonary involvement Arrhythmias Systolic LV dysfunction (late, infrequent) Valvular regurgitation Palpitations Arrhythmias Anginal chest pain LV hypertrophy (increased oxygen demand) Impaired coronary supply (functional hypoperfusion versus fixed stenosis) LV outflow obstruction Syncope Arrhythmias Third-degree AV blocks LV outflow obstruction AV, atrioventricular; LV, left ventricular Reproduced from Linhart et al., Acta Paediatr 2002;91 (Suppl 439):15–20 with permission from Blackwell Publishing Typical electrocardiogram from a patient with Fabry disease PR intervals are short, there is marked left ventricular hypertrophy and prominent ST depressions and T-wave inversions Adapted from Linhart et al., In Mehta, Beck and Sunder-Plassmann, editors. Fabry disease: perspectives from 5 years of FOS. Oxford: Oxford PharmaGenesis Ltd; 2006: p. 189–201 with permission Hypertrophic cardiomyopathy in a patient with Fabry disease Short-axis cine magnetic resonance image showing that the ventricular cavity is virtually absent in systole because of concentric hypertrophy of myocardial fibres From Lidove et al., Am J Roentgenol 2006;186:1184–91. Reprinted with permission from the American Journal of Roentgenology Typical distribution of angiokeratomas on the skin of a patient with Fabry disease Courtesy of Dr Thomas Jansen, Bochum, Germany Cornea verticillata: the most typical ocular sign in Fabry disease Reproduced from Sodi et al., In Mehta, Beck and Sunder-Plassmann, editors. Fabry disease: perspectives from 5 years of FOS. Oxford: Oxford PharmaGenesis Ltd; 2006: p. 249–61 with permission. Fabry cataract: a posterior subcapsular ‘spoke-like’ cataract specific to Fabry disease Reproduced from Sodi et al., In Mehta, Beck and Sunder-Plassmann, editors. Fabry disease: perspectives from 5 years of FOS. Oxford: Oxford PharmaGenesis Ltd; 2006: p. 249–61 with permission Increased vessel tortuosity in the conjunctiva and retina of the eye Conjunctiva Retina Reproduced from Sodi et al., In Mehta, Beck and Sunder-Plassmann, editors. Fabry disease: perspectives from 5 years of FOS. Oxford: Oxford PharmaGenesis Ltd; 2006: p. 249–61 with permission Ear manifestations associated with Fabry disease • Most patients with Fabry disease experience hearing loss at some time during their life. The majority suffer from sensorineural hearing loss, with a minority experiencing mixed or conductive hearing losses [1] • The frequency of sudden hearing loss is elevated compared with the general population [2] • About 85% of male patients over 50 years of age and 75% of female patients over 60 years of age suffer from hearing loss severe enough to justify the use of a hearing aid • Tinnitus is much more frequent in patients with Fabry disease than in the normal population and has also been observed in children [3]. Vertigo has also been reported 1. Hajioff et al., Acta Paediatr 2003;92 (Suppl 443):28–30 2. Germain et al., BMJ Med Genet 2002;3:10 3. Keilmann, Acta Paediatr 2003;92 (Suppl 443): 31–2 Gastrointestinal symptoms of Fabry disease • Gastrointestinal symptoms have been reported in 50–70% of patients with Fabry disease [1, 2] • These include postprandial symptoms such as diarrhoea, abdominal bloating, urgency, flatulence, nausea and vomiting. Patients may also experience a sense of early satiety and epigastric discomfort. • The most commonly reported gastrointestinal symptoms are diarrhoea, with frequent loose bowel motions, and cramping abdominal pain [3] • In many patients, there is an alternating pattern of episodes of diarrhoea and normal or reduced bowel activity/constipation. This alternating pattern is reminiscent of irritable bowel syndrome, as are the symptoms of abdominal discomfort and bloating associated with meals [4] 1. MacDermot et al., J Med Genet 2001;38:750–60 2. MacDermot et al., J Med Genet 2001;38:769–75 3. Dehout et al., J Inherit Metab Dis 2004;27:499–505 4. Cremonini & Talley, Gastroenterol Clin North Am 2005;34:189–204 Impact of Fabry disease on quality of life • The symptoms of Fabry disease may clearly have a significant impact upon quality of life • Hoffmann et al. [1] found that quality of life in 120 patients with Fabry disease enrolled in FOS was significantly lower than in age- and sex-matched normal population • The impact of Fabry disease on quality of life has also been shown to be of comparable severity to that reported by patients with AIDS, and more severe than that reported by patients with Gaucher disease [2] • Psychiatric problems have also been reported, with a high incidence of suicide and depression [3, 4] • The frequency of psychiatric complaints may by as high as 18% [3] 1. Hoffmann et al., J Med Genet 2005;42:247–52 2. Gold et al., Qual Life Res 2002;11:317–27 3. Grewal, Int J Psychiatry Med 1993;23:307–12 4. Wendrich et al., J inherit Metab Dis 2001;24 (Suppl 2):139 Other manifestations of Fabry disease • Obstructive airway disease is a relatively late finding in Fabry disease and can be severe [1] • Many male patients show retarded growth or delayed puberty and sparse, fine facial and body hair. In some families, affected males have a characteristic facial appearance similar to that seen in patients with acromegaly • Many patients have some evidence of musculoskeletal defects • A high prevalence of anaemia has been reported among patients with Fabry disease [2]. This may be of clinical relevance as anaemia is a major risk factor for patients with kidney disease, heart failure or stroke, which are important manifestations of Fabry disease • Hypothyroidism [3], osteopenia [4] and oral–dental abnormalities [5] have also been reported 1. Kariman et al., Am J Med 1978;64:911–12 2. Kleinert et al., Kidney Int 2005;67:1955–60 3. Hauser et al., J Inherit Metab Dis 2005;28:715-22 4. Germain et al., Clin Genet 2005;68:93–5 5. Baccaglini et al., Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2001;92:415–19 Fabry disease with predominant manifestations in a single major organ system • Several cases have been reported of patients with Fabry disease with signs and symptoms mostly confined to the heart or kidney [e.g. 1, 2] • These patients generally have relatively high residual levels of α-galactosidase A activity • In some such cases, however, other organ manifestations have not been excluded [3, 4] 1. Nakao et al., N Engl Med J 1995;333:288–93 2. Ogawa et al., Hum Pathol 1990;21:1067–73 3. Cybulla et al., Am J Kidney Dis 2005;45:e82–9 4. Nakao et al., Kidney Int 2003;64:801–7 Fabry disease in children • In recent years there has been a growing awareness that children show signs and symptoms of Fabry disease. Even at an early age, these manifestations may be severe [1]. Nevertheless, the diagnosis may often be missed • Some of the most common early signs and symptoms of Fabry disease in children are acroparaesthesiae, pain, angiokeratomas and gastrointestinal manifestations [1]. These may be seen in both boys and girls, although the onset is generally 2–5 years later in girls • Other signs and symptoms, such as hypohidrosis, fever, ocular manifestations and tinnitus, may also be observed in young patients. Children may report a reduced tolerance for physical activity • Cardiac and renal involvement can also begin in childhood [1–3]; thus early diagnosis and careful monitoring are necessary 1. Ramaswami et al., Acta Paediatr 2006;95:86–92 2. Meroni et al., Contrib Nephrol 1997;122:178–84 3. Ries et al., Eur J Paed 2003;162:767–72 Fabry disease in women • Traditionally, it was believed that Fabry disease was an X-linked recessive disorder; however, it is now clear that females with this X-linked disease can be severely affected [1–3] • Females who are heterozygous for Fabry disease show a wide range of clinical manifestations • Although some individuals may remain completely asymptomatic and have normal levels of α-galactosidase A, others are as severely affected as hemizygous males [1, 2] • Life expectancy is also reduced in women [1, 3, 4] 1. Mehta et al., Eur J Clin Invest 2004; 34:236–42 3. Deegan et al., J Med Genet 2006;43:347–52 2. Whybra et al., J Inherit Metab Dis 2001;24:715–24 4. MacDermot et al., J Med Genet 2001b;38:769–75 Clinical findings in young male and female patients with Fabry disease Adapted from Ries et al., Eur J Pediatr 2003;162:767–72 with permission. © Springer Multiple hyperintensities in the white matter of the brain in a woman with Fabry disease Axial Sagittal T2-weighted magnetic resonance images of the brain of a 58-year-old woman with Fabry disease showing multiple hyperintensities in the central and subcortical white matter Reproduced from Whybra et al., J Inherit Metab Dis 2001;24:715–24 © SSIEM and Kluwer Academic Publishers, with kind permission from Springer Science and Business Media Trafficking of the enzyme α-galactosidase A to cellular lysosomes ERT, enzyme replacement therapy; M6P, mannose-6-phosphate; TGN, trans-Golgi network Reproduced from Beck & Ries, Fabry disease: clinical manifestations, diagnosis and therapy. Oxford: OCC Europe Ltd; 2001 with permission Replagal PI PRESCRIBING INFORMATION: Replagal (agalsidase alfa) 1 mg/ml concentrate for solution for infusion. Please read Summary of Product Characteristics (SmPC) before prescribing. Presentation: Concentrate solution for intravenous infusion: vials of 3 ml (containing 1 ml of concentrate) and 5 ml (3.5 ml of concentrate). Indication: Replagal (agalsidase alfa) is indicated for long-term enzyme replacement therapy in patients with a confirmed diagnosis of Fabry disease (α-galactosidase A deficiency). Dose: Replagal is administered at a dose of 0.2 mg/kg body weight every other week by intravenous infusion over 40 minutes. No dosage adjustment is necessary in patients with renal impairment. The presence of extensive renal damage (eGFR < 60 ml/min) may limit the renal response to enzyme replacement therapy. No dosage adjustment is recommended in patients on dialysis or postkidney transplantation. Contraindications: Life-threatening hypersensitivity (anaphylactic reaction) to the active substance or any of the excipients. Warnings and precautions: 13.7% of patients treated with Replagal in clinical trials have experienced idiosyncratic infusion-related reactions. Overall, the percentage of infusion-related reactions was significantly lower in females than males. The most common symptoms have been rigors, headache, nausea, pyrexia, flushing and fatigue. Serious infusion reactions have been reported uncommonly; symptoms reported include pyrexia, rigors, tachycardia, urticaria, nausea/vomiting, angioneurotic oedema with throat tightness, stridor and swollen tongue. The onset of infusion-related reactions has generally occurred within the first 2–4 months after initiation of treatment with Replagal although later onset (after 1 year) has been reported as well. If mild or moderate acute infusion reactions occur, medical attention must be sought immediately and appropriate actions instituted. The infusion can be temporarily interrupted (5 to 10 minutes) until symptoms subside and the infusion may then be restarted. Mild and transient effects may not require medical treatment or discontinuation of the infusion. In addition, oral or intravenous pre-treatment with antihistamines and/or corticosteroids, from 1 to 24 hours prior to infusion may prevent subsequent reactions in those cases where symptomatic treatment was required. As with any intravenous protein product, allergic-type hypersensitivity reactions are possible. If severe allergic or anaphylactic-type reactions occur, the administration of Replagal should be discontinued immediately and appropriate treatment initiated. The current medical standards for emergency treatment are to be observed. As with all protein pharmaceutical products, patients may develop IgG antibodies to the protein. A low titre IgG antibody response has been observed in approximately 24% of the male patients treated with Replagal. These IgG antibodies appeared to develop following approximately 3–12 months of treatment. After 12 to 54 months of therapy, 17% of Replagal treated patients were still antibody positive whereas 7% showed evidence for the development of immunologic tolerance, based on the disappearance of IgG antibodies over time. The remaining 76% remained antibody negative throughout. No IgE antibodies have been detected in any patient receiving Replagal. The presence of extensive renal damage may limit the renal response to enzyme replacement therapy, possibly due to underlying irreversible pathological changes. In such cases, the loss of renal function remains within the expected range of the natural progression of disease. Paediatrics: Experience in children is limited. Studies in children (0–6 years old) have not been performed and no dosage regimen can presently be recommended. Limited clinical data in children (7–18 years) do not permit recommendation of an optimal dosage regimen Item code: Re-07-059 presently. As no unexpected safety issues were encountered in a six month study with Replagal administered at 0.2 mg/kg, this dose is suggested for children between 7–18 years. Pregnancy and lactation: Very limited data on pregnancies exposed to Replagal (n = 3) have shown no adverse effects on the mother and newborn child. Animal studies do not indicate direct or indirect harmful effects with respect to pregnancy or embryonal/foetal development when exposed during organogenesis. It is not known whether Replagal is excreted in human milk. Caution should be exercised when prescribing to pregnant or nursing women. Interactions: Replagal should not be co-administered with chloroquine, amiodarone, benoquin or gentamicin since these substances have the potential to inhibit intra-cellular α-galactosidase activity. As α-galactosidase A is itself an enzyme, it would be an unlikely candidate for cytochrome P450 mediated drug–drug interactions. In clinical studies, neuropathic pain medicinal products (such as carbamazepine, phenytoin and gabapentin) were administered concurrently to most patients without any evidence of interaction. Side effects: The most commonly reported undesirable effects were infusion associated reactions, which occurred in 13.7% of patients treated with Replagal in clinical trials. Most undesirable effects were mild to moderate in severity. The following AEs have been reported: peripheral oedema, headache, dizziness, dysgeusia, neuropathic pain, tremor, hypersomnia, hypoesthesia, paraesthesia, parosmia, lacrimation increased, tinnitus, tinnitus aggravated, tachycardia, palpitations, flushing, hypertension, cough, hoarseness, throat tightness, dyspnoea, nasopharyngitis, pharyngitis, throat secretion increased, rhinorrhoea, nausea, diarrhoea , vomiting, abdominal pain/discomfort, acne, erythema, pruritus, rash, livedo reticularis, angioneurotic oedema, urticaria, musculoskeletal discomfort, myalgia, back pain, limb pain, peripheral swelling, arthralgia, joint swelling, sensation of heaviness, rigors pyrexia, pain and discomfort, fatigue, fatigue aggravated, feeling hot, feeling cold, asthenia, chest pain, chest tightness, influenza like illness, injection site rash, malaise, corneal reflex decreased, oxygen saturation decreased. Overdosage: No case of overdose has been reported. Pharmaceutical Precautions: Store in a refrigerator (2°C–8°C). Legal Category: POM. Pack Sizes, Product Licence Numbers and Cost: 1 ml of concentrate for solution for infusion in a 3 ml vial. Pack sizes of 1, 4 or 10 vials. 3.5 ml of concentrate for solution for infusion in a 5 ml vial. Pack sizes of 1, 4 or 10 vials. EU/1/01/189/001-006. Price of £356.85 for 3 ml and £1161.57 for 5 ml vials. Further information available from Product Licence Holder: Shire Human Genetic Therapies AB, Rinkebyvägen 11B, SE 182 36 Danderyd, Sweden. Please read the Summary of Product Characteristics before prescribing. Adverse events should be reported to the Yellow Card Scheme. Information about adverse event reporting via this scheme can be found at www.yellowcard.gov.uk. Adverse events should also be reported to Shire Human Genetic Therapies on +44 (0)1223 422707 or faxed on +44 (0)1223 424666. Date of Preparation: 04 January 2007. Item Code: RePIv040107 Date of preparation: May 2007