Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
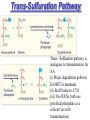
#4 Sulfur Containing Amino Acids 1.Uses of MET/CYS 2.Active Methyl Cycle & Trans-Sulfuration Pathway 3.Defects - Vitamin B6 responsive - Homocystinurea - Cystinurea - Cystinosis Sulfur Containing Amino Acids • 2AA: MET & CYS, MET is converted to CYS through trans-sulfuration pathway • Cystine: dimer of CYS • Homocysteine: not a primary AA Methionine MET - thioether derivative (contains S) - EAA (3.5% animal protein, 1.5% plant protein) IMPORTANCE: (i) as a constituent of P, as well as the initiating AA in P synthesis (ii) source methyl groups for PL, DNA, RNA, hormones via conversion of MET to S-adenosyl MET (iii) source S in CYS via trans-sulfuration pathway (iv) polyamine Cysteine CYS IMPORTANCE: (I) protein synthesis (ii) catalytic site of enzymes where SH is a nucleophile (iii) synthesis of compounds CoA, glutathione, taurine (iv) provides SO4 on complete oxidation CYSTINE: (I) dimer of CYS (ii) formed through oxidation mediated by O2, Cu2+, Mn2+ or Fe2+ (iii) little free in cells (iv) important in disulfide bond formation in intact proteins to provide bridges and stabilize conformation of proteins. Synthesis of CYS/Metabolism of MET 2 pathways: Active Methyl Cycle and the TransSulfuration Pathway. Active Methyl Cycle MET SAM SAH H – Cys Cys First step: Activation of MET → SAM (a high energy compound) Most Rxns with SAM are Methyl transferases Methyl transferred SAM → several compounds where Methyl linked to O or N in acceptor molecule eg, Methylation RNA, DNA, cathecholamines adrenaline synthesis creatine phosphatidyl choline Second step: Free energy loss: SAM → SAH SAH: no longer has a charged sulfonium atom (thioether MET), rxn irreversible This represents a branch point in MET metabolism (direction depends on physiological needs of organism). (I) if MET limited homoCYS remethylated to MET (closed pathway of active methyl cycle) (ii) If CYS needed and SAM adequate then homoCYS trans-sulfuration pathway. (ii) If both CYS and MET are adequate, then other pathways Active Methyl Cycle (i) MET is an EAA (ii) But mammals can synthesize MET from homoCYS if available (can replace MET in diet) because it is the “homoCYS” that cannot be made (iii) Can transfer a methyl group back to homo CYS from betaine or (primarily) 5 methyl THF (tetrahydrofolate) Therefore regenerate MET P synthesis, methylation and CYS synthesis (iv) Crossroads for 2 important vitamins : folic acid and vitamin B12 (Vitamin B12 deficiency folate deficient state) (v) One of the major determinants whether homoCYS MET is level of 5 methyl THF. SAM blocks 5 methyl THF production therefore decreased MET production when [SAM] increases Trans-Sulfuration Pathway Trans - Sulfuration pathway is analagous to transamination for AA (i) Major degradation pathway for MET in mammals (ii) End Product is CYS (iii) Two RXNs, both use pyridoxal phosphate as a cofactor (as with transamination) FATE OF ATOMS OF MET CH3 S methyl transfer CH2 converted to propionyl CoA succinyl CoA CH2 CH— NH3 COO- CYS NH4+ CO2 glucose Inborn Errors of Sulfur - containing AA Metabolism (i) Two of these defects - homocystinuria (type I) & cystathioninuria involve enzymes of trans-sulfuration pathway (homo Cys Cys) (ii) Homocystinuria (2nd most common genetic AA disease) named due to homocystine urine (CYS dimer) homocystine& MET & homoCYS blood which spills over into urine. Dimer (homocystine) forms spontaneously in tissue (iii) 4 types of homoCYS ( see table) Type I = defect in cystathionine synthase (enzyme homoCYS CYS: first step) Also with less homoCYS MET therefore increase homoCYS Type II: defect in methylene THF reductase decrease [5 methyl THF] which is necessary to methylate homoCYS Type III: Decrease in B12 Type IV: malabsorption of B12 Type I Homocystinuria B6 responsive - (responsive to vitamin therapy) - 50% Type I - doses B6 up to 1 g / day Remember defect in cystathionine synthase enzyme that converts homoCYS CYS (Step I) pyridoxal phosphate is a cofactor and that B6 Ppal Km mutants: do not bind cofactor as avidly therefore need much more cofactor to increase enzyme activity Note: overly large dose (4-5 g / day) nervous system dysfunction. Type I Homocystinuria B6 unresponsive - enzyme mutation that does not involve cofactor binding site therefore dietary therapy (i) add betaine Why? Enhance alternate pathway (ii) keep MET (use plant P like soybean/lentil which has half the MET of animal P) (iii) CYS conditionally essential because MET cannot be converted to CYS (iv) Start therapy early due to serious clinical symptoms Symptoms - Mental defects - Skeletal malformations (osteoporosis) due to defective collagen formation (homoCYS interferes with corsslinking collagen) -Dislocation ocular lens(abnormal lens ligaments) - Thromboembolism and vascular occlusion (decreased life expectancy) with damage to lining of blood vessels major cause morbidity and mortality. Note:Increased homoCYS is a recognized risk factor for heart attack / stroke in adults TREATMENT:Dietary folic acid which decreases homoCYS (via trans-sulfuration pathway) Cystathioninuria - much rarer than Type I - due to a defect in cystathion(in)ase (Step 2 CYS synthesis) - less clinical abnormalities than Type I - accumulation cystathionine in blood/urine (not detectable normally) - responds to B6 supplementation in some cases (B6 Ppal cofactor to enzyme) which also suggests a Km mutant #4 Sulfur-Containing Amino Acids: Homocystinuria Case Discussion A 6-year-old girl was brought to the hospital with vision problems. She was found to have a downward dislocation of the left lens. Her mother indicated that the girl’s birth was normal, but that she lagged in development. She was unable to crawl until 1-year-old and did not walk until 2 years. Speaking was also delayed. She had long, thin bones; on roentgenographic examination the lower femur showed signs of osteoporosis. An older brother had similar symptoms, but had been diagnosed as having Marfan’s syndrome. A simple cyanide-nitroprusside test of the patient’s urine was positive, suggesting homocystinuria. This was confirmed by amino acid analysis of the plasma, which revealed homocystine, an abnormally high methionine level, and other sulfurcontaining compounds that were derivatives of homocysteine. The patient was treated with a low-methionine diet supplemented with folic acid and pyridoxine. Symptoms / clinical characteristics 1. Downward dislocation left lens Slow development - crawling / walking / speaking. Osteoporosis, long thin bones 2. Cyanide/nitro prusside urine test = +ve 3. Plasma AA homoCYS, MET, homoCYS derivatives Suggests that homoCYS due to decrease conversion to CYS. MET also therefore no problem in conversion homoCYS MET Discussion 1. What is the origin of the homocystine excreted in this disease? 2. What are some of the metabolic substances formed by the enzymatic reactions that use S-adenosylmethionine as the methylating agent? 1. Origin of homocystine: dimer of homoCYS, product of excess homoCYS, overflows into urine 2. These include RNA, DNA, amino terminal groups P’s, precursors of melatonin, creatine, epinephrine, phosphatidylcholine, methyl cobalamin (B6). Discussion 3. What are some causes of homocystinuria in humans? 4. How would one test for a deficiency of cystationine βsynthase in this patient? 3. Any part of pathway that impedes MET homoCYS pathway homoCYS - decrease cystathionine B synthase (Step 1 for CYS) - decreased syn of MET from homoCYS (5 MeTHF or B12 or enzyme ) genetic or nutritional - Also 6 azauridine administration: anticancer agent which inh PPal enzymes therefore decreases both step 1 and 2 - bacterial action on cystathionine in urine 4. Test of enzyme: Measure enzyme in cells (human skin fibroblasts)to define hetero or complete mutation. Test addition of pyridoxal phosphate in vitro since with effectiveness of therapy. (ie B6 sensitivity) Discussion 5. Explain why pyridoxine is useful in the treatment of some patients with homocystinuria. 6. What effect would a diet low in folate have on this patient? 5. (i) pyridoxine and/or B6 improvement in behaviour and IQ (ii) Diet decreased MET, add smaller meals to prevent MET overload (iii) add Vitamin B12 and folic acid 6. Diet decrease folate: Good or Bad? BAD Seriously effect 5 MeTHF rxn therefore homoCYS even worse (no alternate pathway) Discussion 7. What might account for the homocystinuria of an apparently normal infant (not in this case) with severe megaloblastic anemia and who was exclusively breast-fed by a strict vegetarian mother? 8. Describe the genetics of homocystinuria (cystathionine βsynthase deficiency). 7. HomoCYS B12 deficiency - vegetarians: no eggs, no dairy, scrubbed vegetables decrease cobalamin therefore not enough vitamin B12 synthesize cofactor - therefore no homoCYS MET Can result in permanent neurological damage 8. Autosomal Recessive (1:45,000), Defects in gene chromosome 21, Heterogeneous