Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
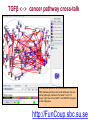
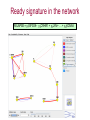
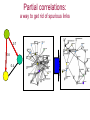
Expression signatures as biomarkers: solving combinatorial problems with gene networks Andrey Alexeyenko Department of Medical Epidemiology and Biostatistics, Karolinska Institute FunCoup is a data integration framework to discover functional coupling in eukaryotic proteomes with data from model organisms Bmouse Human Rat Fly Yeast High-throughput evidence ? Find orthologs Amouse Andrey Alexeyenko and Erik L.L. Sonnhammer. Global networks of functional coupling in eukaryotes from comprehensive data integration. Genome Research. Published in Advance February 25, 2009 FunCoup • • • • Each piece of data is evaluated Data FROM many eukaryotes (7) Practical maximum of data sources (>50) Predicted networks FOR a number of eukaryotes (10…) • Organism-specific efficient and robust Bayesian frameworks • Orthology-based information transfer and phylogenetic profiling • Networks predicted for different types of functional coupling (metabolic, signaling etc.) http://FunCoup.sbc.su.se TGFβ <-> cancer pathway cross-talk FunCoup was queried for any links between members of TGFβ pathway (left blue circle) and habituées of known cancer pathways (members of at least 7 out of 18 groups; right blue circle). MAPK1 and MAPK3 belonged to both categories. http://FunCoup.sbc.su.se FunCoup: recapitulation of known cancer pathways Figure 5 from: The Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008 Sep 4. [Epub ahead of print] The same genes submitted to FunCoup No TCGA data were used. Outgoing links are not shown. × Outcome, Optimal treatment, Severity/urgency etc. Single molecular markers are (often) far from perfect. Combinations (signatures) should perform better. The problem: How to select optimal combinations? Biomarker discovery in network context The idea: Construct multi-gene predictors with regard to network context • Reduce the computational complexity • Make marker sets biologically sound Accounting for network context is taking either: a) network neighbors or b) genes at remote network positions Procedure “Rotterdam” dataset (Wang et al., 2005): 286 patients Clinical data: Expression: ~22000 probes × Estrogen receptor status: +/ – Lymph. node status: all – Relapse : yes/no and time (days) Individual probe p-values (~22000): Estrogen receptor-specific ability to predict relapse Select most significant probes (1000): Candidate members for marker signatures Compile set of probes: N probes at a time (e.g. N=20 or N=50) 1. Split data: 75% to train, 25% to test. 2. Produce a linear regression equation (weight terms step-wise, reward for performance, penalize for complexity) on the train sub-set. 3. Apply the equation to the test set to predict outcome (relapse yes/no). Repeat m times4. Record the specificity/sensitivity (Type I/II error rates) as ROC curve. RELAPSE = γ1g1 + γ2g2 + γ3g3 + … + γNgN Procedure Select most significant probes (1000): Candidate members for marker signatures Compile set of probes: N probes at a time (e.g. N=20 or N=50) Test X randomly retieved sets Take the best ones Account for the network context 1. Split data: 75% to train, 25% to test. 2. Produce a linear regression equation (weight terms step-wise, reward for performance, penalize for complexity) on the train sub-set. 3. Apply the equation to the test set to predict outcome (relapse yes/no). Repeat m times4. Record the specificity/sensitivity (Type I/II error rates) as ROC curve. RELAPSE = γ1g1 + γ2g2 + γ3g3 + … + γNgN Candidate signature in the network Biomarker candidates Ready signature in the network RELAPSE = γ1EIF3S9+ γ2CRHR1 + γ3LYN + … + γNKCNA5 Testing “top”, “free”, and “network” approaches Estrogen receptor status:positive Frequency netw free Top Estrogen receptor status:negative netw free 91% 92% 93% 94% Quality of prognosisrelapse/no relapse Top 95% 96% 97% (area under ROC curve) Frequency 90% 93% 94% 95% 96% 97% Quality of prognosisrelapse/no relapse 98% 99% (area under ROC curve) Signature involves genes mutated in cancer Cancer individuality: each tumor is unique in its molecular state and set of mutated/disordered genes Tumour tcga-02-0114-01a-01w Partial correlations: a way to get rid of spurious links 0.7 0.6 0.4 Cancer individuality via network view Functional coupling transcription ? transcription transcription ? methylation methylation ? methylation mutation methylation mutation transcription mutation ? mutation + mutated gene is a framework for biomarker discovery: • Markers can be discovered and presented in the network dimension. • Choice of data types to incorporate is unlimited – from metabolite profiling to patient phenotypes. Useful features: • Web-based resource ready for further expansion and presenting new research results in an interactome perspective; • Cross-species network comparison of human and model organisms. • Efficient query system to retrieve network environments of interest. http://FunCoup.sbc.su.se Thank you for attention! Decomposing biological context Develomental Common rPLC = 0.95 rPLC = 0.88 ANOVA (Analysis Of VAriance): Look at F-ratios: Signal of interest / Residual (“error”) variance Dioxin-enabled rPLC = 0.76 Accounting for edge features: dioxin-enabled vs. dioxin-sensitive links Andrey Alexeyenko, Deena M Wassenberg, Edward K Lobenhofer, Jerry Yen, Erik LL Sonnhammer, Elwood Linney, Joel N Meyer Transcriptional response to dioxin in the interactome of developing zebrafish. submitted. a