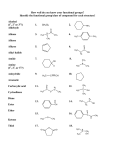
Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Structure Determination for Organic Compounds Structure determination is used for identification of an unknown compound and for elucidation of the structure for a novel compound. Vast numbers of possible structures exist (several million organic compounds have been synthesized or isolated), and therefore determination of the structure is often demanding. Methods for structure determination 1. Use many different chemical reactions with known results 2. Chromatographic methods 3. Spectroscopic methods – especially IR and NMR 4. Mass Spectrometry Spectroscopy involves measuring the interaction of molecules with electromagnetic radiation. Essentially the entire electromagnetic spectrum, from γ radiation to radio waves, can be used for studying molecules. Wavelength Range (meters) Photon energy (kJ/mol) Spectroscopic region Spectroscopic technique 10–13 109 γ-radiation Mössbauer 10–10 106 X-radiation X-ray diffraction, X-ray emission spectroscopy 10–7 1200 Far ultraviolet Far UV spectroscopy 3 x 10–7 (300 nm) 430 Near UV UV/Vis spectroscopy 4 x 10–7 to 7 x 10–7 300 to 170 Visible UV/Vis spectroscopy 10–6 to 10–4 120 to 1.2 Infrared IR spectroscopy Microwave Electron paramagnetic resonance Radio Nuclear magnetic resonance 0.01 1 0.01 0.0001 Wavelength is inversely proportional to energy. E = hν = € Copyright © 2003 Mark Brandt, Ph.D. 1 hc λ IR spectroscopy uses radiation at wavelengths somewhat longer than visible light. IR uses relatively low energy (λ= 1 µm corresponds to 120 kJ/mol, which is considerably less than covalent bond strength). IR is an absorbance technique. Both single beam and dual beam instruments have been designed; FTIR is usually single beam, using computer subtraction of baseline. IR measures vibrational modes. Rotational modes and other effects broaden the quantized absorbance from lines to peaks of varying widths. Can have two-atom stretch and bend. Can have three-atom stretch (symmetric and asymmetric) and bend. Can have larger number of atoms involved in the modes as well; most of the multi-atom modes are weak bands. IR absorption requires a change in the dipole of the molecule as a result of the vibrational mode, with larger dipole changes resulting in more intense bands. Intensity is proportional to: 1. The number of molecules present in IR beam (which means that both higher concentrations and thicker films result in more intense bands) 2. The amount of a particular bond-type in the molecule (for example, the C-H stretch is usually intense, because most organic compounds have many C-H bonds) 3. The polarity of the bond (more polar bonds result in more intense IR bands). IR spectrum most commonly used extends from ~2.5 to ~16 µm (~4000 cm-1 to ~500 cm-1). Wavenumber (cm-1) is a frequency measurement wavenumbers = number of wave peaks in a centimeter. Some older sources use “microns”, which are wavelength units (cm-1 = 10,000 / microns) A form of Hooke’s Law describes the position of the IR absorption bands: wavenumbers = ν = 1 m + m2 k 1 2πc m1m2 where k is a force constant proportional to bond strength, and m1 and m2 are the masses of the atoms involved. Light atoms have high frequencies; strong bonds € result in high frequencies. For organic compounds, the IR spectrum is comprised of four regions: 1. Hydrogen region (4000 – 2700 cm-1) 2. Triple-bond region (2300 – 2000 cm-1) 3. Double-bond region (1900 – 1500 cm-1) 4. Fingerprint region (1500 – 500 cm-1) Copyright © 2003 Mark Brandt, Ph.D. 2 The spectrum from 3800 – 2700 cm-1 is the “hydrogen region”: essentially all of the bands are due to hydrogen stretching modes. Bond strengths (and therefore frequencies) generally follow the trends: O-H > N-H > C-H, and C H > C H > C H The C-H stretching bands are: C H ~3300 cm-1 C H ~3070 cm-1 C H 3000-2850 cm-1 The –C-H stretch is rarely above 3000 cm-1, so stretching bands for hydrogens bonded to sp2 and sp hybridized carbons tend to stand out from the –C-H peak. The –C-H peak is almost always a multiplet, because of symmetric and asymmetric stretching modes. Aromatic rings usually have more than one C-H stretch band (usually two or three) from 3150 to 3050 (note that aromatic carbons are sp2 hybridized). A final type of C-H stretch occurs in aldehydes; these compounds have two peaks (~2800 cm-1 and ~2700 cm-1) due to stretching modes for the carbonyl-hydrogen bond. Alcohols have the O-H stretch, which is observed at about 3300 cm-1. (Note that the O-H stretch is affected by hydrogen bonding, which weakens and broadens the band; in the gas phase, or in dilute solutions, the O-H stretch occurs at ~3600 cm-1.) The O-H stretch of a carboxylic acid is normally very broad due to hydrogenbonding; the main peak is normally at 3100 to 3000, with the C-H stretching bands showing through the peak. Primary and secondary amines and amides have N-H stretching bands at 34003250; the bands are sharper and less intense than O-H stretching bands. Primary amines and amides have doublets, while secondary compounds have single bands. The N-H stretching peaks often extend out from a broad band that probably is due to hydrogen bonding of the hydrogens. Note that tertiary amines and lack an N-H, and therefore lack the stretching band. The “triple-bond region” is 2300-2000 cm-1, with few other groups absorbing in this region. C C 2150-2100 cm-1 C N 2260-2220 cm-1 Copyright © 2003 Mark Brandt, Ph.D. 3 Note, however, that terminal alkynes usually exhibit the stretch band, but internal triple bonds usually absorb weakly at most due to the limited dipole moment change during stretching. The “double-bond region” is 1900-1500 cm-1, although a few non-double bond groups also absorb in this region. The most useful band in the double-bond region is the carbonyl C=O stretch at ~1710 cm-1. Careful analysis of the exact position of the carbonyl stretch can yield information about the atoms that surround the carbonyl because different groups alter the strength of the C=O bond. Esters and 5-membered ring ketones have slightly higher frequency (~30 wavenumber increase); conjugation to a double bond or aromatic ring or nitrogen (i.e. amide) lowers the frequency by a similar amount). A halogen bonded to the carbonyl raises the frequency by ~80 cm-1. The C=C stretch is usually present at ~1650, but it is usually a weak band. In addition, the structure around the double bond (especially conjugation) may alter the position of the peak. Nitro groups have the “walrus teeth”: sharp, strong peaks at ~1550 cm-1 and ~1400 cm-1 (the values are shifted to smaller wavenumbers when conjugated). The paired peaks (these are the symmetric and asymmetric stretching modes for the N-O 1.5 bond) are usually visible in spite of their appearance in the middle of the fingerprint region because of their strong intensity and similar size. Aromatics have a series of bending modes that are typically quite strong at 14 to 15 microns (600-800 cm-1). These are difficult to see. Aromatic rings also have up to four bands, usually of similar intensity, at 1600, 1580, 1500, and 1450 cm-1. Although these bands are buried in the fingerprint region, they may act as confirmation of aromatic character deduced from other features in the spectrum. Finally, aromatic compounds have a series of relatively weak overtone bands at 1800 – 2000 cm-1 and little else is usually present in this frequency range. Using many scans on FTIR, it is frequently possible to pick out these aromatic overtones; different substitution patterns result in different patterns, so it is often possible to determine the number and position of substitutents on benzene ring. The handout contains spectra that show the different peak arrangements observed for different substitution patterns. The “fingerprint region” (below 1500 cm-1) consists of single bond stretches for non-hydrogen atoms, and most of the bending vibrational modes. Bands in this region are much more difficult to interpret; however, comparison of the fingerprint region of an experimental spectrum with a reference spectrum for a known compound may confirm the identification of the unknown. C-O single bond stretch, C-N single bond stretch, and C-C single bond stretch are usually in the fingerprint region (900-1200 cm-1). C-C stretching bands are almost always weak, because the bond is not very polar. Copyright © 2003 Mark Brandt, Ph.D. 4 Alcohols have a C-O stretch, which is somewhat variable (generally at 1100-1400 cm-1), depending on whether the compound is a primary, secondary or tertiary alcohol, with the strength and the position of the C-O stretch dependent on the connectivity of the carbon. The C-O stretch is often difficult to distinguish from among the other bands in the fingerprint region, and is much less useful in assessing the functional group than other spectral features present in the molecule. Esters also have a C-O stretch at 1300-1000, which is usually rather strong. Ethers are difficult to identify; except for a C-O stretch near 1150 cm-1, ethers look like alkanes. Stretching bands are higher energy than bending bands. For most molecules, bending modes are difficult to interpret. Most are present in the fingerprint region below ~1500 cm-1. The most useful bending band is the methyl group “umbrella bend”, which is normally found at ~1380 cm-1. This appears as a single band for isolated methyl groups, and as a double for isopropyl groups. In assessing a spectrum, look at all of the features. Remember that impurities add bands to the spectrum. Look for the intense signals first. Do not try to interpret every last peak in the spectrum. Copyright © 2003 Mark Brandt, Ph.D. 5