Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
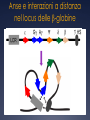
genomica comparata L’analisi comparata dei genomi analizza cosa c’è in comune e cosa c’è di unico tra specie diverse a livello genomico (anatomia comparata molecolare?) può costituire il modo più sicuro ed affidabile per identificare geni (Genscan, Orpheus ecc.) e predire le loro funzioni e interazioni ad esempio, la funzione di alcuni geni o sequenze di DNA umane può essere chiarita studiando le loro controparti in organismi più semplici Aree di Pertinenza della Genomica Comparata la genomica comparata include il confronto di: • tutte le proteine note o predette (proteomi), • la posizione dei geni nei genomi, • il numero e la posizione dei repeats, un’altra area della genomica comparata comprende: • l’analisi della diversità intra-specifica (SNPs, variabilità dei microsatelliti e dei livelli di espressione genica) • l’associazione tra queste variazioni e la risposta all’ambiente e/o alle malattie Cosa si può fare con la Genomica Comparata si può confrontare la localizzazione relativa di geni correlati in uno stesso genoma e in genomi separati come di ogni gruppo locale di geni che possa avere un significato funzionale o regolativo gli ortologhi sono geni così conservati in genomi diversi che si predice che le proteine codificate abbiano la stessa struttura e funzione e che si siano generati a partire da un antenato comune in seguito a speciazione Identificazione di geni “ortologhi” per identificare gli ortologhi, ogni genedell’organismo 1 viene usata come query in una ricerca di similarità contro tutti i geni dell’organismo 2 in prima approssimazione, si assume che il gene del genoma 2 più simile al gene “query” ne sia l’ortologo Genoma 2 Genoma 1 query Confronto di interi genomi per aumentare l’affidabilità della predizione, si richiede che per ogni coppia di ortologhi, ognuno dei due geni risulti quello più simile all’altroquando confrontato con l’intero genoma Genoma 1 Genoma2 se entrambe le relazioni sono vere ==> le due proteine gialle si possono proporre come ortologhe Similitudine e Posizione in genere si richiedono anche bassi E-value (< 10-20) e che l’allineamento includa almeno il 60-80% della sequenza query (per evitare di incappare in paraloghi) in organismi correlati, in genere si trovano conservati sia il contenuto in geni che l’ordine dei geni nei genomi la similarità di sequenza e di posizione confermano eventualmente l’ortologia per organismi via via meno correlati, gruppi di geni locali restano correlati, ma riarrangiamenti cromosomici possono spostare i cluster in altre posizioni sul genoma Pathway metabolici nei procarioti nei genomi dei microorganismi, i geni di uno stesso pathway metabolico possono sono contigui in quanto coregolati trascrizionalmente in un operone o posti sotto il controllo di uno stesso promotore Pathway metabolici negli eucarioti anche negli eucarioti, geni correlati possono ritrovarsi in cluster se ne deduce che, nota la funzione di geni contigui, la funzione di un gene può esserne inferita per facilitare il confronto tra genomi, è necessario proporre un vocabolario comune (Gene Ontology) confronto intra- genoma • identifica famiglie geniche (distingue geni unici da geni appartenenti a famiglie geniche) • identifica paraloghi (coppie di geni simili possono essere paraloghi) confronto tra genomi • identifica ortologhi • identifica famiglie geniche • identifica domini Confronto Intra-Genoma il confronto INTRA-genoma aiuta ad evitare casi di errata valutazione di ortologia tra i Es.=> X (genoma A) è ortologo di Y (genoma B) ? Potrebbe invece essere che Y e Z siano paraloghi, e che X sia ortologo di Z Genoma A X Genoma B ? Y Z Definiamo un grafo come una struttura matematica destinata a rappresentare una relazione binaria tra elementi di uno o più insiemi possiamo utilizzare dei grafi per descrivere relazioni binarie (per esempio di similarità) tra proteine di uno stesso proteoma e spostare l’ottica del confronto dalla coppia al cluster Tools for Comparative Genomics UCSC Browser: This site contains the reference sequence and • working draft assemblies for a large collection of genomes. The Ensembl project produces genome databases • Ensembl: for vertebrates and other eukaryotic species, and makes this information freely available online. The Map Viewer provides a wide variety of genome • MapView: mapping and sequencing data.[26] A comprehensive suite of programs and databases for • VISTA: comparative analysis of genomic sequences. It was built to visualize the results of comparative analysis based on DNA alignments. The presentation of comparative data generated by VISTA can easily suit both small and large scale of data. Bacteria Genomes Alignments This alignment of eight Yersinia bacteria genomes reveals 78 locally collinear blocks conserved among all eight taxa. Evolution of the FOXP2 gene Human FOXP2 gene and evolutionary conservation is shown in and multiple alignment (at bottom of figure) in this image from the UCSC Genome Browser. Conservation tends to cluster around exons. Gene Ontology (GO) PRODOTTO GENICO FUNZIONE MOLECOLARE PROCESSO BIOLOGICO COMPONENTE CELLULARE Un prodotto genico può avere una o più funzioni molecolari, partecipare a uno o più processi biologici, e essere associato ad uno o più componenti cellulari. (DAG) Le categorie indicate in rosso sono rappresentate con una frequenza significativamente (p < 0.01) più elevata di quella attesa. COMPARATIVE GENOMICS AT THE VERTEBRATE EXTREMES Dario Boffelli, Marcelo A. Nobrega and Edward M. Rubin NATURE REVIEWS | GENETICS VOLUME 5 | JUNE 2004 | 457 Annotators of the human genome are increasingly exploiting comparisons with genomes at both the distal and proximal evolutionary edges of the vertebrate tree. Despite the sequence similarity between primates, comparisons among members of this clade are beginning to identify primate- as well as human-specific functional elements. At the distal evolutionary extreme, comparing the human genome to that of non-mammal vertebrates such as fish has proved to be a powerful filter to prioritize sequences that most probably have significant functional activity in all vertebrates. Human–Fugu rubripes conserved non-coding sequences (CNS) in the human genome 51 human–F. rubripes CNSs (in purple). One cluster of human–F. rubripes CNS in more detail. DACH is involved in embryonic development some of the non-coding sequences conserved in humans and F. rubripes act as enhancers in mouse embryos. In this assay, the sequence being tested is cloned upstream of a β-galactosidase reporter gene. If the cloned sequence is an enhancer, it will activate the reporter gene, which can be detected in an assay that stains the tissues that express β-galactosidase (in blue). Extreme conservation in enhancers shared by human and fish A core enhancer in an intron in DACH is >98% identical for 350 bp in humans, mice and rats. In the ~1 billion years of parallel evolutionary time that separates human, mouse, rat, chicken, frog and fish, only 6 substitutions occurred in a 120-bp fragment that corresponds to an enhancer, 4 of which occurred in the frog lineage alone, and none occurred in the mammalian lineage. Sonic hedgehog expression in the limbs is regulated by an enhancer at a distance of 1 Mb a | Human–Fugu rubripes sequence comparisons, generated by VISTA, identify a conserved non-coding sequence in intron 5 of LMBR1 (red box), which drives the expression of a reporter gene in a pattern that resembles the expression of sonic hedgehog (SHH) (arrows in b). Insertional mutagenesis in this region in mice results in preaxial polydactly (arrows in c). In humans, mutations in this enhancer are also associated with preaxial polydactyly (arrows in d). *Between mammals and fish. The molecular function and biological process of each gene were obtained from the Gene Ontology Phylogenetic shadowing analyses sequence variation in a multiple alignment identifies regions that accumulate variation at a slower rate Phylogenetic Generally, positions with several sequence differences in multiple branches of the phylogenetic tree are more likely to be evolving at a fast rate, and in turn identify the least variable regions shadowing analyses sequence variation in a multiple alignment to identify regions that accumulate variation at a slower rate. Each position in the multiple alignment is fitted to a phylogenetic model to calculate the likelihood that the position is evolving at a fast or a slow rate . The slowly evolving regions often correspond to functional The slowly evolving sequences. regions often correspond to functional sequences. The use of highly similar sequences minimizes ambiguity in the computation of the multiple alignment. Moreover, the phylogenetic tree that relates the data is easy to infer and facilitates the comparative assembly of draft sequence from nonhuman primates to the reference human genome. Identification of adaptively evolving genes d /d >1 N S can result from a decrease in population size or a relaxation of selection. The ratio of non-synonymous to synonymous substitutions, dN/dS, indicates the type of selection that a gene is subject to. An excess of non-synonymous substitutions in pairwise sequence comparisons (d /d >1) indicates that 1 of the 2 sequences is S undergoing positive selection. To N determine in which lineage positive selection occurred, however, the sequence of a third species is needed. Defining functional DNA elements in the human genome Manolis Kellis et al. PNAS April 29, 2014 | vol. 111 | no. 17 | 6131–6138 With the completion of the human genome sequence, attention turned to identifying and annotating its functional DNA elements. As a complement to genetic and comparative genomics approaches, the Encyclopedia of DNA Elements Project was launched to contribute maps of RNA transcripts, transcriptional regulator binding sites, and chromatin states in many cell types. The resulting genome-wide data reveal sites of biochemical activity with high positional resolution and cell type specificity that facilitate studies of gene regulation and interpretation of noncoding variants associated with human disease. However, the biochemically active regions cover a much larger fraction of the genome than do evolutionarily conserved regions, raising the question of whether nonconserved but biochemically active regions are truly functional. Here, we review the strengths and limitations of biochemical, evolutionary, and genetic approaches for defining functional DNA segments, potential sources for the observed differences in estimated genomic coverage, and the biological implications of these discrepancies. We also analyze the relationship between signal intensity, genomic coverage, and evolutionary conservation. Our results reinforce the principle that each approach provides complementary information and that we need to use combinations of all three to elucidate genome function in human biology and disease. The complementary nature of evolutionary, biochemical and genetic evidence Encyclo pedia of DNA Elements (ENCODE) Project DNA that produces a phenotype upon alteration GERP++ elements from 34 mammal alignments Human genome coverage by ENCODE fragments per kilobase of exon per million reads (FPKM) RNA-seq Genomic footprint / ChIP-seq Relationship between ENCODE signals and conservation only 5% of mammalian genomes are under strong evolutionary constraint across multiple species At present, we cannot distinguish which low-abundance transcripts are functional Regions with higher signals generally exhibit higher levels of evolutionarily conservation Epigenetic and evolutionary signals in cisregulatory modules (CRMs) of the HBB complex Anse e interazioni a distanza nel locus delle -globine Lo “switch” fetale-adulto nel locus delle -globine Il locus Albumina / Alfa-fetoproteina (ALB/AFP) Hind III (AAGCTT) 13.4 5 10 13.5 45.9 46.3 47.8 48.2 15 20 25 30 35 40 50 45 55 60 III II ALB Prom inattive ALB Prom AFP + AFP - ALB - ALB + III II Ealb Eafp Before birth Eafp AFP Prom AFP Prom ACTIVE AFP Prom inactive III II Ealb Eafp ALB Prom After birth ACTIVE x Ealb x Kb Sau3A (GATC) BASTA !!