Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
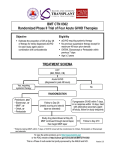
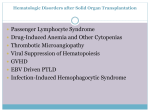
hematology Board Review Manual Statement of Editorial Purpose The Hospital Physician Hematology Board Review Manual is a study guide for fellows and prac ticing physicians preparing for board exami nations in hematology. Each manual reviews a topic essential to the current practice of hematology. PUBLISHING STAFF PRESIDENT, Group PUBLISHER Bruce M. White Acute and Chronic GraftVersus-Host Disease Series Editor: Eric D. Jacobsen, MD Instructor of Medicine Harvard Medical School Attending Physician Dana-Farber Cancer Institute Boston, MA editorial director Debra Dreger Associate EDITOR Rita E. Gould assistant EDITOR Farrawh Charles Contributor: Corey Cutler, MD, MPH, FRCP(C) Assistant Professor of Medicine Harvard Medical School Boston, MA executive vice president Barbara T. White executive director of operations Jean M. Gaul PRODUCTION Director Suzanne S. Banish PRODUCTION assistant Table of Contents Nadja V. Frist ADVERTISING/PROJECT director Patricia Payne Castle sales & marketing manager Deborah D. Chavis Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2 Acute GVHD. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2 Chronic GVHD. . . . . . . . . . . . . . . . . . . . . . . . . . . 6 NOTE FROM THE PUBLISHER: This publication has been developed with out involvement of or review by the Amer ican Board of Internal Medicine. References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11 Cover Illustration by Kathryn K. Johnson Copyright 2008, Turner White Communications, Inc., Strafford Avenue, Suite 220, Wayne, PA 19087-3391, www.turner-white.com. All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, mechanical, electronic, photocopying, recording, or otherwise, without the prior written permission of Turner White Communications. The preparation and distribution of this publication are supported by sponsorship subject to written agreements that stipulate and ensure the editorial independence of Turner White Communications. Turner White Communications retains full control over the design and production of all published materials, including selection of topics and preparation of editorial content. The authors are solely responsible for substantive content. Statements expressed reflect the views of the authors and not necessarily the opinions or policies of Turner White Communications. Turner White Communications accepts no responsibility for statements made by authors and will not be liable for any errors of omission or inaccuracies. Information contained within this publication should not be used as a substitute for clinical judgment. www.turner-white.comHematology Volume 3, Part 1 Hematology Board Review Manual Acute and Chronic Graft-Versus-Host Disease Corey Cutler, MD, MPH, FRCP(C) INTRODUCTION Graft-versus-host disease (GVHD) is a common complication of allogeneic hematopoietic stem cell transplantation (HSCT) that occurs when donor T cells attack the tissues of an immunocompromised host. There are 2 distinct syndromes of GVHD: acute GVHD and chronic GVHD. Acute GVHD remains one of the most significant barriers to successful allogeneic HSCT, accounting for a substantial portion of early transplantrelated morbidity and mortality. Clinically relevant acute GVHD occurs in 35% to 40% of patients transplanted from a matched sibling donor1 and in 40% to more than 50% of recipients of unrelated donor grafts.2,3 Severe acute GVHD occurs in up to 20% of recipients of related donors1 and up to 35% of unrelated donors.2,3 Likewise, chronic GVHD has become increasingly more common as the proportion of patients who become long-term survivors of HSCT increases. It is estimated that up to 50% to 70% of long-term survivors of allogeneic HSCT will have some manifestations of chronic GVHD. Chronic GVHD and its complications are the most frequent cause of late (> 2 yr) death after transplantation, underscoring its importance.4,5 Although there is an association between chronic GVHD and a protective graft-versus-tumor effect, the morbidity and mortality associated with chronic GVHD remains substantial. Chronic GVHD is also associated with important limitations on patient quality of life. Using an illustrative case, this review discusses the pathophysiology, risk factors, clinical features, and management of both acute and chronic GVHD. ACUTE GVHD CASE PRESENTATION A 43-year-old man is referred for allogeneic HSCT. He was previously diagnosed with acute myelogenous leukemia with normal cytogenetics. He received induction chemotherapy followed by consolidation and attained a complete remission. Fifteen months after the initial diagnosis, the patient suffers a relapse. He re- Hospital Physician Board Review Manual ceives reinduction chemotherapy and attains a second remission. The patient has a healthy, human leukocyte antigen (HLA)-matched sibling who will donate stem cells, but he is concerned about the risk of developing acute GVHD. • What is the pathophysiology of acute GVHD? Acute GVHD results from the complex interaction of donor T cells and host tissues that involves recognition of major and minor histocompatibility antigens in an inflammatory milieu. Critical factors that modulate the alloreactivity seen in acute GVHD include donor–host tolerance mechanisms and the judicious use of immune suppression. The pathophysiology of acute GVHD involves both the innate and adaptive immune systems and is thought to follow a reproducible pattern of (1) tissue damage from conditioning regimen, (2) donor T-cell activation, and (3) an inflammatory effector phase.6 • What are the risks for developing acute GVHD? Several risk factors can predict the occurrence of acute GVHD (Table 1). These factors can be separated into distinct categories, of which only some may be selectively modulated by clinical decision making. For example, many of the donor-recipient factors often cannot be altered, particularly when only a single donor is available for stem cell donation (eg, sex matching). However, risk factors related to the graft composition and the choice of the conditioning regimen often can be selected by the transplant team at the time of transplantation. • How is acute GVHD prevented? ACUTE GVHD PROPHYLAXIS Without prophylaxis, acute GVHD would be nearly universal; therefore, some form of prophylaxis is always employed in the peritransplant period. Two main strategies are used in preventing GVHD: graft manipulation and pharmacologic prophylaxis. Graft Manipulation Consistent with the notion that GVHD is induced by donor T cells coinfused with the stem cell graft, graft manipulation to remove T cells (T-cell depletion [TCD]) is www.turner-white.com A c u t e a n d C h ro n i c G r a f t - Ve r s u s - H o s t D i s e a s e one effective method available for preventing GVHD.7 Graft manipulation can be accomplished using either in vivo or ex vivo approaches. Either approach can reduce T-cell numbers in the stem cell graft by several logfold. In vivo TCD methods use horse- or rabbit-derived antithymocyte globulin or alemtuzumab to immunologically purge T cells. In vivo TCD simultaneously eliminates both recipient T cells that could mediate graft rejection and donor T cells that could mediate acute GVHD. However, other cellular components such as B cells, natural killer cells, and dendritic cells of both donor and host origin are likely to be affected by these polyspecific antibodies. The specific depletion of these cellular components may or may not influence the risk of acute GVHD. Ex vivo TCD can be accomplished via several methods. Physical separation of T cells from the stem cells can be performed using methods such as lectin agglutination (using sheep red blood cell rosetting) or counterflow centrifugal elutriation in a closed ex vivo system. An alternative strategy is the use of magnetic bead columns coated with monoclonal antibodies. With this method, either positive selection of CD34+ progenitors, with subsequent elutriation of the captured cells from the column for transplantation, or negative selection of T cells using monoclonal T-cell antibodies can be performed. The resulting eluate that passes through the column is then used for transplantation.2 Studies in both matched related and unrelated transplantation have demonstrated that TCD can be an effective strategy to prevent acute GVHD, with rates of acute GVHD as low as 10% in matched related transplantation8 and 34% to 38% in matched unrelated donor transplantation.9 In a recent prospective, multicenter, randomized phase II/III trial that enrolled 405 patients who underwent unrelated donor marrow transplantation, the incidence of acute GVHD was significantly lower in the ex vivo TCD arm as compared with the conventional (pharmacologic) GVHD prophylaxis arm (39% versus 63%; P < 0.0001). However, overall disease-free survival at 3 years was 30%, with no difference between the TCD and non-TCD arms (27% versus 34%; P = 0.16).10 The lack of a survival advantage seen in this and other trials has been explained by impaired posttransplant immune reconstitution, which causes an increased incidence of infections as well as an increased risk of graft failure, an increase in the risk of posttransplant Epstein-Barr virus– associated lymphoproliferative disorders, and a reduction in graft-versus-tumor activity, leading to increased relapse rates.2,5 Despite these limitations and the lack of contemporary randomized evidence supporting their use, in vivo TCD strategies are commonly used as GVHD prophylaxis. However, the continued use of these agents Table 1. Risk Factors for Acute GVHD Factor Condition That Increases Risk of Acute GVHD Donor-recipient factors Major HLA disparity (HLA class I, II) HLA mismatched donor > matched donor Minor HLA disparity (mHA) Unrelated donor > related donor Sex matching Mismatch > match Donor parity Multiparity > nulliparity Donor age Older donor > younger donor ABO type ABO mismatch > ABO match Donor CMV serostatus CMV positive > CMV negative Cytokine gene poly morphisms Numerous associated with acute GVHD Stem cell graft factors Stem cell source PBSC > BM > UCB Graft composition Higher CD34+ cell count > lower CD34+ cell count* Higher T-cell dose > lower T-cell dose* Transplantation factors Conditioning intensity Myeloablative > reduced-intensity regimens BM = bone marrow; CMV = cytomegalovirus; GVHD = graft-versushost disease; HLA = human leukocyte antigen; mHA = minor histocom patibility antigens that are expressed in the context of HLA by antigen presenting cells; PBSC = peripheral blood stem cell; UCB = umbilical cord blood. *Controversial. relates more to their role in preventing chronic GVHD, particularly in alternative donor transplantation, rather than as prophylaxis of acute GVHD.11,12 Pharmacologic Prophylaxis Pharmacologic prevention of acute GVHD is practiced more commonly in North America than prophylaxis with TCD. Immunosuppressive drugs are used to reduce the alloimmune activation of donor T cells. Methotrexate, which impairs purine synthesis in T cells and thus prevents T-cell expansion in response to neo antigen, was the first agent used to prevent GVHD. Single-agent methotrexate was supplanted by cyclosporine when it became widely available. Cyclosporine inhibits interleukin-2–mediated T-cell expansion via inhibition of calcineurin. Studies comparing cyclosporine monotherapy with the combination of cyclosporine and methotrexate demonstrated superiority of the 2-drug regimen over monotherapy, and the current standard was established.13 Newer agents have been developed to prevent acute GVHD. The most prominent of these is tacrolimus, which is used interchangeably with cyclosporine. In www.turner-white.comHematology Volume 3, Part 1 A c u t e a n d C h ro n i c G r a f t - Ve r s u s - H o s t D i s e a s e contrast to cyclosporine, tacrolimus does not interact with calcineurin or its downstream effectors, but both compounds share a final common pathway of T-cell inhibition. Large phase III studies comparing tacrolimus and methotrexate with cyclosporine and methotrexate after matched related and unrelated transplantation have been performed. In the matched related donor setting, 329 patients were randomized to receive either tacrolimus with methotrexate or cyclosporine and methotrexate. The incidence of grade II through grade IV acute GVHD was 31.9% in the tacrolimus arm and 44.4% in the cyclosporine arm.14 Similarly, the incidence of grade II through grade IV acute GVHD was 56% among 46 patients randomized to tacrolimus versus 74% among 63 patients randomized to receive cyclosporine in the unrelated donor study.15 Other investigational but promising strategies to prevent acute GVHD involve newer immunosuppressants, such as sirolimus16 or mycophenolate mofetil,17 as well as agents with activity in the treatment of advanced established acute GVHD. CASE CONTINUED The patient undergoes transplantation from his HLA-matched sibling. He receives cyclophosphamide and total body irradiation as conditioning therapy, and tacrolimus with methotrexate as GVHD prophylaxis. Twenty-three days after successful transplantation, the patient presents to his physician complaining of a new rash and profuse watery diarrhea. A complete metabolic panel also reveals significant elevation of alanine aminotransferase, aspartate aminotransferase, and total bilirubin levels. • What are the clinical features of acute GVHD? CLINICAL FEATURES OF ACUTE GVHD Acute GVHD is a clinicopathologic syndrome involving the skin, liver, and gut. The median time to the diagnosis of acute GVHD varies with conditioning, with recipients of high-dose therapy and transplantation being diagnosed at a median of 17 days,18 as compared with recipients of reduced-intensity conditioning transplantation being diagnosed at a median of 3 months.19 The skin is the most commonly affected organ in acute GVHD. In a representative study that enrolled 160 pa tients who underwent HLA-matched HSCT, over 75% of patients who developed acute GVHD had some cutaneous involvement and 44% had skin involvement as their only manifestation of acute GVHD.20 The initial manifestation of acute GVHD is most commonly a maculopapular exanthema. The rash may be pruritic or painful and red to violaceous in color. Typically, a rash Hospital Physician Board Review Manual that involves the palms and soles suggests acute GVHD, most likely due to the concentration of hematopoietic stem cells in the rete ridges. The presence of rash at these locations helps differentiate the rash caused by acute GVHD from a rash caused by drug eruption, as the latter generally spares these areas. As the rash intensifies, confluent involvement of the cheeks, ears, neck, and trunk is noted. In its most severe form, bullae formation may occur with epidermal necrosis and desquamation, mimicking toxic epidermal necrolysis. The gastrointestinal (GI) tract is the second most commonly involved organ in acute GVHD, with up to half of individuals being affected in 1 study.20 In general, signs of enteric GVHD develop as the chemoradiotherapy effects resolve following the first several weeks after transplantation; however, these syndromes may overlap, making an early diagnosis difficult. Symptoms of acute GVHD of the small bowel and colon include profuse watery diarrhea, intestinal bleeding, crampy abdominal pain, and, in its most severe form, paralytic ileus. The diarrhea is often green, mucoid, watery, and mixed with exfoliated cells, forming fecal casts. Some patients may develop a variant of GVHD that involves the upper GI tract and is associated with anorexia, nausea, and dyspepsia; these patients may not manifest lower tract involvement. Endoscopic findings of enteric GVHD range from normal to extensive edema and mucosal sloughing. Lesions may be most prominent in the cecum, ileum, and ascending colon but may also involve the stomach, duodenum, and rectum. Endoscopy and biopsy are recommended for the diagnosis of lower GI tract in order to exclude other diagnoses. The liver is the least commonly affected organ in acute GVHD, with less than 20% of patients with acute GVHD having some degree of hepatic involvement.20 Cholestatic jaundice is the most common manifestation; however, a concomitant or isolated transaminitis is not uncommon. In severe cases, hepatic failure with encephalopathy may occur. Percutaneous or transjugular liver biopsy is recommended for the diagnosis of hepatic GVHD, when feasible, as there are no imaging studies that reliably can differentiate hepatic GVHD from other causes of liver dysfunction, such as drug toxicity or veno-occlusive disease of the liver. In general, biopsies of the target organs are often obtained to confirm the diagnosis of acute GVHD. However, the diagnosis remains largely clinical and therapy often must be initiated before the results of biopsies are known. Once the diagnosis is obtained, the severity of acute GVHD can be staged (Table 2). The modified Seattle-Glucksberg criteria, also called the Consensus criteria, are used to stage acute GVHD.21,22 www.turner-white.com A c u t e a n d C h ro n i c G r a f t - Ve r s u s - H o s t D i s e a s e Table 2. Grading and Staging of Acute Graft-Versus-Host Disease Individual Organ Staging Organ Skin Liver Gut Stage BSA (%)* Bilirubin (mg/dL) Diarrhea (mL/day) 1 Rash < 25 2–2.9 500–1000 or biopsy-proven upper GI involvement 2 Rash 25–50 3–6 1000–1500 3 Rash > 50 6.1–15 1500–2000 4 Generalized erythroderma with bullae 15 > 2000 or severe abdominal pain with or without ileus Overall Grade Skin Liver Gut I Stage 1–2 None None II Stage 3 Stage 1 Stage 1 II — Stage 2–3 Stage 2–4 IV Stage 4 Stage 4 — Overall Grade Skin Liver Gut A Stage 1 None None B Stage 2 Stage 1 or 2 Stage 1 or 2 C Stage 3 Stage 3 Stage 3 D Stage 4 Stage 4 Stage 4 Consensus Grading IBMTR Grading BSA = body surface area; GI = gastrointestinal; IBMTR = International Bone Marrow Transplant Registry. *Use “Rule-of-Nines” to determine BSA. (Data from Glucksberg et al,21 Przepiorka et al,22 and Rowlings et al.23) In this system, each individual organ is given a stage, and a composite grade is calculated. A more recent staging system proposed by the International Bone Marrow Transplant Registry uses the same individual organ staging but modifies the overall grading scheme slightly.23 Both systems predict overall survival from acute GVHD equally well. • How is acute GVHD treated and what are the outcomes of this treatment? TREATMENT AND OUTCOME OF ACUTE GVHD Once established, acute GVHD must be treated aggressively, unless it is limited to a low-surface area skin rash alone (stage I or II skin disease). If left untreated, some of these rashes will resolve spontaneously or will resolve with local application of corticosteroid creams. Corticosteroids are lympholytic and inhibit inflammatory cytokine cascades; this class of drugs is considered standard primary systemic therapy for acute GVHD. The dose and route of administration of corticosteroids may vary slightly among transplant centers, but the recommended initial dose of corticosteroids for moderate to severe (grade II–IV) acute GVHD is 2 mg/kg/day of methylprednisolone; randomized trials have not demonstrated an advantage for using higher doses.24 Large retrospective reviews have shown that the response rate to single-agent corticosteroid therapy is approximately 50%.18 It is important to note that the pace of response to primary therapy varies with the organs involved with GVHD. Although erythematous cutaneous acute GVHD may show improvement within 24 hours after initiating therapy, it is unlikely that GI or hepatic GVHD would respond within the same timeframe. Systemic therapy for acute GVHD is continued for at least 1 to 2 weeks after a complete remission is attained and then a gradual taper is initiated. The pace of the steroid taper should generally be no greater than a 10% reduction per week, but this recommendation is affected by factors such as the durability of the response to corticosteroids and the toxicity of high-dose steroid administration. Despite an initial response, many patients will experience a flare of their acute GVHD upon steroid taper, requiring reinstitution of higher doses of corticosteroids or the addition of other agents. In acute GVHD developing after matched unrelated and www.turner-white.comHematology Volume 3, Part 1 A c u t e a n d C h ro n i c G r a f t - Ve r s u s - H o s t D i s e a s e Table 3. Therapeutic Agents Used for Steroid-Refractory Acute GVHD T-cell immunosuppression IL-2 mediated Cyclosporine, tacrolimus mTOR mediated Sirolimus Antimetabolite/ chemotherapy Mycophenolate mofetil, methotrexate, pentostatin Biologic anti–T-cell therapy Polyclonal antibodies Antithymocyte globulin Monoclonal antibodies Muromonab-CD3, visilizumab,* ABX-CBL,* daclizumab, inolimomab, basiliximab, alemtuzumab, alefacept Immunotoxin/conjugate Denileukin diftitox TNF-α blockade Infliximab, etanercept Phototherapy PUVA, extracorporeal photopheresis Cellular therapy Mesenchymal stem cells Topical/directed therapy Oral beclomethasone diproprionate or budesonide for intestinal GVHD; intra-arterial steroid or methotrex ate infusion GVHD = graft-versus-host disease; IL = interleukin; mTOR = mam malian target of rapamycin; PUVA = psoralen plus ultraviolet A; TNF = tumor necrosis factor. *Not commercially available. related donor transplantation, durable remission of acute GVHD with steroid alone were reported in only 21% and 40% of patients, respectively.25,26 The suboptimal response and long-term survival associated with acute GVHD treated with corticosteroids alone has prompted the investigation of additional systemic immunosuppressive agents in the initial therapy of acute GVHD. Unfortunately, this strategy has generally been unsuccessful, as the benefit of more effective initial GVHD control is often offset by a greater risk of infection and other side effects, thus resulting in no net survival benefit.27,28 Currently, there is no established consensus for de fining failure of primary therapy in acute GVHD. A set of criteria common to some clinical trials define steroid-refractory acute GVHD as (1) progression after 3 days, (2) no change after 5 to 7 days, or (3) incomplete response after 14 days of steroid treatment with methylprednisolone at a dose of 2 mg/kg/day or equivalent.29 For patients with steroid-refractory GVHD, there is no clear standard for salvage or second-line treatment, and survival remains poor despite seemingly high response rates to certain second-line therapeutic agents. Table 3 provides a summary of therapeutic agents available for the treatment of steroid-refractory acute GVHD. In addition to immunosuppressive therapy, supportive care is critical for achieving a favorable outcome in steroidrefractory acute GVHD. Organ-specific supportive mea- Hospital Physician Board Review Manual sures for the skin involve using a topical emollient and meticulous wound care, which may include admission to a burn unit. For the GI tract, bowel rest and intravenous hyperalimentation are required. Antimotility agents are employed but may not be clinically useful, as GI GVHD is a secretory process. Octreotide or other somatostatin analogues may be of benefit in controlling the secretory component of GI GVHD.30,31 Ursodeoxycholic acid may be considered as a supportive measure for hepatic GVHD. Given that most patients with steroid-refractory GVHD will succumb to infection, systematic monitoring and judicious use of antibiotic prophylaxis is paramount. Standard infection prophylaxis in patients with GVHD should include trimethoprim-sulfamethoxazole or equivalent agents to prevent Pneumocystis jirovecii pneumonia, acyclovir to prevent herpesvirus reactivation, and prophylaxis against invasive fungal infections. Monitoring for viral pathogens such as cytomegalovirus and fungal pathogens using serum assays for β-glucan and galactomannan is required. CASE CONTINUED A presumptive diagnosis of acute GVHD is made, the patient is rehospitalized, and corticosteroids (2 mg/kg/day) are instituted. Bowel rest is prescribed. After 3 days of therapy, the patient’s skin rash improves. At 10 days of therapy, his liver function tests have normalized and his diarrhea is resolved. The patient is discharged on a tapering course of corticosteroids. CHRONIC GVHD CASE CONTINUED Eight months after transplantation, the patient returns for routine follow-up. He has been successfully weaned off all immunosuppressive therapy, and there is no evidence of leukemic relapse. He now notes the progressive onset of dry and itchy eyes, oral ulcers, and a new rash over his torso. His complete blood count reveals eosinophilia and thrombocytopenia with a platelet count of 80,000/µL (normal, 150,000–350,000/µL). A presumptive diagnosis of extensive chronic GVHD is made, and corticosteroids and a calcineurin inhibitor are initiated. • What are the risk factors for developing chronic GVHD? Risk factors for chronic GVHD are similar to those for acute GVHD, with the contribution of the stem cell source and disparities among major and minor HLA disparities being the most prominent risks. The recent www.turner-white.com A c u t e a n d C h ro n i c G r a f t - Ve r s u s - H o s t D i s e a s e administration of a donor lymphocyte infusion is a risk factor, and the prior occurrence of acute GVHD is also a well-established risk factor. • What is the pathophysiology of chronic GVHD? Unlike acute GVHD, the pathophysiology of chronic GVHD is poorly understood. Human chronic GVHD shares several characteristics with systemic autoimmune diseases such as scleroderma, Sjögren’s syndrome, primary biliary cirrhosis, and systemic lupus erythematosus, which helps relate the pathophysiology of this disorder to other autoimmune (or in the case of chronic GVHD, alloimmune) disorders. T cells play a critical role in GVHD and graft-versus-leukemia immune responses after allogeneic HSCT. Depending on the model system utilized, both CD4+ and CD8+ T cells have been implicated as primary mediators of chronic GVHD, and there are data supporting the role of other subsets of T cells, including CD4+CD25+FoxP3+ regulatory T cells, as well as the subcategories of central and effector memory T cells. Dendritic cells certainly play a role as antigen presenting cells, and most recently, B cells have been implicated in the pathophysiology of chronic GVHD.32 • What are the clinical features of chronic GVHD? • How is chronic GVHD diagnosed? CLINICAL FEATURES The diagnosis of chronic GVHD is largely clinical. The signs and symptoms of chronic GVHD are detailed in Table 4. A detailed physical examination with clinical testing of potentially affected organs is crucial. Laboratory and radiologic testing of affected organs may also provide diagnostic utility. Common supplemental clinical tests that readily can be performed include Schirmer’s test for ocular dryness, portable spirometry for the diagnosis of bronchiolitis, and hand-held dynamometry for musculoskeletal involvement. These tests are also useful for monitoring patient with active chronic GVHD. Traditionally, only GVHD features that occurred beyond 100 days from transplantation were considered part of the spectrum of chronic GVHD. It is now clear that chronic GVHD may present prior to day 100, and features of late acute GVHD may present well after day 100. Thus, it is the syndrome’s clinical features, not its timing, that determines the diagnosis. Recently, a National Institutes of Health (NIH) Con sensus Project outlined the clinical features seen in chronic GVHD. The diagnosis of chronic GVHD is currently based on satisfying a number of clinical criteria as set forth by the Consensus committee (Table 4).29 The diagnosis of chronic GVHD requires (1) the distinction of the clinical syndrome from acute GVHD; (2) the presence of at least 1 diagnostic clinical sign of chronic GVHD or presence of at least 1 distinctive manifestation confirmed by pertinent biopsy or other relevant tests; and (3) exclusion of other possible diagnoses.33 • How is chronic GVHD staged? STAGING CHRONIC GVHD Traditionally, chronic GVHD was staged using a method initially developed from a small cohort of patients treated in Seattle.34 In this system, chronic GVHD patients are divided into 2 categories—clinically limited or clinically extensive disease. Limited chronic GVHD is comprised of patients with localized skin dysfunction or hepatic dysfunction. All others are then considered to have clinically extensive chronic GVHD. Although easy to remember, this system simply groups patients by their requirement for systemic therapy and is of little clinical utility today. As such, the NIH Consensus committee proposed a more complicated but more clinically meaningful scoring system, with the goal of standardizing patient staging for clinical trial design and interpretation. This new system incorporates both the number of target organs involved as well as the severity of the organ involvement.33 The utility of this new system in predicting response to therapy or long-term outcome has not yet been validated in prospective clinical trials. The individual organ scoring system is found in the Figure. The summation of the individual organ scores is then used to place the chronic GVHD patient into 1 of 3 categories: mild, moderate, or severe (Table 5). • How is chronic GVHD treated? TREATMENT Single-agent corticosteroids are the mainstay of therapy for chronic GVHD, and other agents such as cyclosporine, while active, are associated with a low response rate when used alone. Instituting combination therapy as initial therapy for chronic GVHD has not been shown to be beneficial, with the exception of a reduction in steroidassociated side effects. In a trial that compared combination cyclosporine and prednisone with single-agent prednisone as initial therapy for chronic GVHD, the requirement for secondary immunosuppressive therapy due to a lack of response or an insufficient response was no different between study arms. In addition, no differences in transplant-related mortality, relapse rate, or overall mortality were noted. The median duration of therapy with corticosteroids and cyclosporine for chronic GVHD was 1.6 years, and despite dual immunosuppressive www.turner-white.comHematology Volume 3, Part 1 A c u t e a n d C h ro n i c G r a f t - Ve r s u s - H o s t D i s e a s e Table 4. Signs and Symptoms of Chronic Graft-Versus-Host Disease (GVHD) Organ or Site Skin Diagnostic (Sufficient to Establish the Diagnosis of Chronic GVHD) Distinctive (Seen in Chronic GVHD, but Insufficient Alone to Establish a Diagnosis of Chronic GVHD) Other Features* Poikiloderma Lichen planus-like features Sclerotic features Morphea-like features Lichen sclerosis-like fea tures Depigmentation Nails Dystrophy Longitudinal ridging, splitting, or brittle features Onycholysis Pterygium unguis Nail loss (usually symmetric; affects most nails)† Scalp and body hair New onset of scarring or non scarring scalp alopecia (after recovery from chemoradio therapy) Scaling, papulosquamous lesions Mouth Lichen-type features Hyperkeratotic plaques Restriction of mouth open ing from sclerosis Eyes Lichen planus-like features Vaginal scarring or stenosis GI tract Esophageal web Strictures or stenosis in the upper to mid third of the esophagus† Sweat impairment Ichthyosis Keratosis pilaris Hypopigmentation Hyperpigmentation Thinning scalp hair, typically patchy, coarse, or dull (not explained by endocrine or other causes) Premature gray hair Gingivitis Mucositis Erythema Pain Photophobia Periorbital hyperpigmentation Blepharitis (erythema of the eyelids with edema) Confluent areas of punctate kera topathy Erosions† Fissures† Ulcers† Exocrine pancreatic insuf ficiency Liver Lung Muscles, fascia, joints Erythema Maculopapular rash Pruritus Xerostomia Mucocele Mucosal atrophy Pseudomembranes† Ulcers† New onset of dry, gritty, or pain ful eyes‡ Cicatricial conjunctivitis Keratoconjunctivitis sicca‡ Genitalia Common (Seen with Both Acute and Chronic GVHD) Anorexia Nausea Vomiting Diarrhea Weight loss Failure to thrive (infants and children) Total bilirubin, alkaline phos phatase > 2× ULN† ALT or AST > 2× ULN† Bronchiolitis obliterans diag Bronchiolitis obliterans diagnosed nosed with lung biopsy with PFTs and radiology‡ Fasciitis Myositis or polymyositis‡ Edema Joint stiffness or con Muscle cramps tractures secondary to Arthralgia or arthritis sclerosis Bronchiolitis obliterans organizing pneumonia (continued) Hospital Physician Board Review Manual www.turner-white.com A c u t e a n d C h ro n i c G r a f t - Ve r s u s - H o s t D i s e a s e Table 4. Signs and Symptoms of Chronic GVHD (continued) Organ or Site Diagnostic (Sufficient to Establish the Diagnosis of Chronic GVHD) Distinctive (Seen in Chronic GVHD, but Insufficient Alone to Establish a Diagnosis of Chronic GVHD) Other Features* Hematopoietic and immune Thrombocytopenia Eosinophilia Lymphopenia Hypo- or hypergammaglobu linemia Autoantibodies (AIHA and ITP) Other Pericardial or pleural effusions Ascites Peripheral neuropathy Nephrotic syndrome Myasthenia gravis Cardiac conduction abnormal ity or cardiomyopathy Common (Seen with Both Acute and Chronic GVHD) AIHA = autoimmune hemolytic anemia; ALT = alanine aminotransferase; AST = aspartate aminotransferase; GI = gastrointestinal; ITP = idiopathic thrombocytopenic purpura; PFTs = pulmonary function tests; ULN = upper limit of normal. *Can be acknowledged as part of the chronic GVHD symptomatology if the diagnosis is confirmed. †In all cases, infection, drug effects, malignancy, or other causes must be excluded. ‡Diagnosis of chronic GVHD requires biopsy or radiology confirmation (or Schirmer’s test for eyes). (Adapted from Filipovich AH, Weisdorf D, Pavletic S, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant 2005;11:948–9. Copyright 2005, with permission from Elsevier.) therapy, only 54% of patients were successfully weaned from immunosuppressive medications at 5 years. Mortality directly attributable to chronic GVHD was 17% in the combination immunosuppressive arm.35 Trials that included additional agents (eg, thalidomide) as part of initial therapy for chronic GVHD also did not demonstrate an advantage.35,36 Typical doses of corticosteroids used in the initial management of chronic GVHD range from 0.5 to 1.0 mg/kg/day of prednisone. Some investigators prefer to administer corticosteroids on alternate days to reduce the incidence of steroid-related side effects, since therapy with corticosteroids is often very prolonged. In contrast to the tapering of steroids in acute GVHD, most investigators prefer to maintain steroid doses at therapeutic levels for up to 3 months once a complete remission has been attained before initiating a taper. Currently, there is no standard second-line therapy for chronic GVHD when primary therapy is insufficient. As a result, therapy often consists of prolonged administration of corticosteroids in conjunction with other immunosuppressive medications. The interested reader is referred to these recent publication for a detailed discussion of regimens used in chronic GVHD.37,38 Response rates to individual agents vary, with responses noted in up to 75% of patients treated with mycophenolate mofetil. Up to 20% of these patients will attain a complete response to this agent. Use of novel agents (eg, sirolimus, thalidomide) as well as newer approaches to treating chronic GVHD (eg, rituximab, extracorporeal phototherapy) has also been explored. In addition to systemic therapy for chronic GVHD, organ-specific therapy is required. This organ-specific therapy can be vital and includes the topical administration of immunosuppressants to individual organs (ie, corticosteroid-containing oral rinses, calcineurin inhibitor ocular preparations) and other supportive care measures to maintain the integrity of the affected organ. A complete guide to supportive care of individual organs for patients with chronic GVHD was also produced by the NIH Consensus Group.39 • What is the prognosis for patients with chronic GVHD? PROGNOSIS There have been numerous attempts to define the prognosis of chronic GVHD. Several factors have repeatedly demonstrated adverse prognostic importance, such as a progressive onset of chronic GVHD arising out of acute GVHD and thrombocytopenia at the time of diagnosis. Others, such as poor performance status at the time of diagnosis, hyperbilirubinemia, and extensive skin involvement, have not retained prognostic significance across studies.4,40 CASE CONCLUSION Nine months after initiating corticosteroids and www.turner-white.comHematology Volume 3, Part 1 A c u t e a n d C h ro n i c G r a f t - Ve r s u s - H o s t D i s e a s e SCORE 0 PERFORMANCE SCORE: KPS ECOG LPS ❑A symptomatic and fully active (ECOG 0; KPS or LPS 100%) SCORE 1 ❑ Symptomatic, fully ambulatory, restricted only in physically stren uous activity (ECOG 1, KPS or LPS 80%–90%) SCORE 2 SCORE 3 ❑ Symptomatic, ambulato ❑ Symptomatic, limited self-care, ry, capable of self-care, > 50% of waking hours in bed > 50% of waking hours (ECOG 3–4, KPS or LPS < 60%) out of bed (ECOG 2, KPS or LPS 60%–70%) SKIN ❑ No symptoms Clinical features: ❑ Maculopapular rash ❑ Lichen planus-like features ❑P apulosquamous lesions or ichthyosis ❑ Hyperpigmentation ❑ Hypopigmentation ❑ Keratosis pilaris ❑ Erythema ❑ Erythroderma ❑ Poikiloderma ❑ Sclerotic features ❑ Pruritus ❑ Hair involvement ❑ Nail involvement % BSA involved ❑ < 18% BSA with disease ❑ 19%–50% BSA OR ❑ > 50% BSA OR deep sclerotic signs but NO sclerotic involvement with super features “hidebound” (unable features ficial sclerotic features to pinch) OR impaired mobility, “not hidebound” (able ulceration, or severe pruritus to pinch) MOUTH ❑ No symptoms ❑ Mild symptoms with disease signs but not limiting oral intake sig nificantly ❑ Moderate symptoms with disease signs with partial limitation of oral intake ❑ Severe symptoms with disease signs on examination with major limitation of oral intake EYES Mean tear test (mm): ❑ >10 ❑ 6–10 ❑≤5 ❑ Not done ❑ No symptoms ❑ Mild dry eye symptoms not affecting ADL (requiring eyedrops ≤ 3× daily) OR asymp tomatic signs of kerato conjunctivitis sicca ❑ Moderate dry eye symptoms partially affecting ADL (requir ing drops > 3× daily or punctal plugs), WITHOUT vision impairment ❑ Severe dry eye symptoms signifi cantly affecting ADL (special eye ware to relieve pain) OR unable to work because of ocular symp toms OR loss of vision caused by keratoconjunctivitis sicca GI TRACT ❑ No symptoms ❑ Symptoms such as dys ❑ Symptoms associated phagia, anorexia, nausea, with mild to moderate vomiting, abdominal pain weight loss (5%–15%) or diarrhea without significant weight loss (< 5%) ❑ Symptoms associated with signifi cant weight loss > 15%, requires nutritional supplement for most calorie needs OR esophageal dilation LIVER ❑ Normal LFT ❑ Elevated bilirubin, AP,* AST or ALT < 2× ULN ❑B ilirubin > 3 mg/dL or bilirubin, enzymes 2– 5× ULN ❑ Bilirubin or enzymes > 5× ULN LUNGS† ❑N o symptoms ❑ Mild symptoms (short ness of breath after climbing 1 flight of steps) ❑ Moderate symptoms (shortness of breath after walking on flat ground) ❑ Severe symptoms (shortness of breath at rest; requiring 02) ❑ F EV1 > 80% OR LFS = 2 ❑ FEV1 60%–79% OR LFS 3–5 ❑ FEV1 40%–59% OR LFS 6–9 ❑ FEV1 ≤ 39% OR LFS 10–12 FEV1 DLCO (continued) 10 Hospital Physician Board Review Manual www.turner-white.com A c u t e a n d C h ro n i c G r a f t - Ve r s u s - H o s t D i s e a s e SCORE 0 SCORE 1 SCORE 2 SCORE 3 JOINTS AND FASCIA ❑N o symptoms ❑ Mild tightness of arms or legs, normal or mild decreased ROM AND not affecting ADL GENITAL TRACT ❑N o symptoms ❑ Symptomatic with mild ❑ Symptomatic with mod ❑ Symptomatic WITH advanced signs on exam AND erate signs on exam signs (stricture, labial agglutina no effect on coitus and AND with mild dys tion or severe ulceration) AND minimal discomfort with pareunia or discomfort severe pain with coitus or inabil gynecologic exam with gynecologic exam ity to insert vaginal speculum ❑ Tightness of arms or ❑ Contractures WITH significant legs OR joint contrac decrease of ROM AND signifi tures, erythema thought cant limitation of ADL (unable due to fasciitis, moder to tie shoes, button shirts, dress ate decrease ROM self etc.) AND mild to moderate limitation of ADL Other indicators, clinical manifestations or complications related to chronic GVHD (check all that apply and assign a score to its severity (0–3) based on its functional impact where applicable (none = 0; mild –1; moderate –2; severe –3) Esophageal stricture or web___ Ascites (serositis)___ Myasthenia gravis___ Polymyositis___ Platelets < 100,000/µL ___ Pericardial effusion___ Nephrotic syndrome___ Cardiomyopathy___ Cardiac conduction defects___ Progressive onset___ Pleural effusion(s)___ Peripheral neuropathy___ Eosinophilia > 500 µL___ Coronary artery involvement___ OTHERS: Specify: _____________________________________________________________________________________________________ Figure. Individual Organ Scoring Criteria for Chronic GVHD. ADL = activities of daily living; ALT = alanine aminotransferase; AP = alkaline phosphatase; AST = aspartate aminotransferase; BSA = body surface area; DLCO = diffusion capacity of carbon monoxide; ECOG = Eastern Cooperative Oncology Group; FEV1 = forced expiratory volume in 1 second; GI = gastrointestinal; GVHD = graft-versus-host disease; KPS = Karnofsky Performance Status; LFS = Lung Function Score; LFT = liver function test; LPS = Lansky Performance Status; PFT = pulmonary function testing; ROM = range of motion; ULN = upper limit of normal. *AP may be elevated in growing children and is not reflective of liver dysfunction. †Pulmonary scoring should be performed using both the symptom and PFT scale whenever possible. When discrepancy exists between pulmonary symptom and the PFT scores, the higher value should be used for final scoring. Scoring using the LFS is preferred, but if DLCO is not available, grading using FEV1 should be used. The percent predicted FEV1 and DLCO (adjusted for hematocrit but not alveolar volume) should be converted to a numeric score as follows: ≥ 80% = 1; 70%–79% = 2; 60%–69% = 3; 50%–59% = 4; 40%–49% = 5; < 40% = 6. The LFS + FEV1 score = DLCO score, with a possible range of 2–12. (Adapted from Filipovich AH, Weisdorf D, Pavletic S, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant 2005;11:952–3. Copyright 2005, with permission from Elsevier.) tacrolimus, all signs and symptoms of chronic GVHD have resolved. The patient remains on a tapering course of corticosteroids and a therapeutic dose of tacrolimus. He has developed glucose intolerance and requires oral hypoglycemic medications. His hematologic parameters have returned to normal, and routine staging with bone marrow examination has revealed no evidence of recurrence of leukemia. REFERENCES 1. Champlin RE, Schmitz N, Horowitz MM, et al. Blood stem cells compared with bone marrow as a source of hematopoietic cells for allogeneic transplantation. IBMTR Histocompatibility and Stem Cell Sources Working Committee and the European Group for Blood and Marrow Transplanation (EBMT). Blood 2000;95:3702–9. Table 5. Definitions of Mild, Moderate, or Severe Chronic Graft-Versus-Host Disease (GVHD) Mild chronic GVHD Up to 2 organ (excluding lung) scores = 1 Moderate chronic GVHD 1 organ (excluding lung) score = 2 or Three or more organ scores = 1 or Lung score = 1 Severe chronic GVHD Lung score = 2 or Any other organ score = 3 Data from Filipovich AH, Weisdorf D, Pavletic S, et al. National Insti tutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant 2005;11:948–9. www.turner-white.comHematology Volume 3, Part 1 11 A c u t e a n d C h ro n i c G r a f t - Ve r s u s - H o s t D i s e a s e 2. Castro-Malaspina H, Harris RE, Gajewski J, et al. Unrelated donor marrow transplantation for myelodysplastic syndromes: outcome analysis in 510 transplants facilitated by the National Marrow Donor Program. Blood 2002; 99:1943–51. 3. McGlave PB, Shu XO, Wen W, et al. Unrelated donor marrow transplantation for chronic myelogenous leukemia: 9 years’ experience of the national marrow donor program. Blood 2000;95:2219–25. 4. Lee SJ, Klein JP, Barrett AJ, et al. Severity of chronic graft-versus-host disease: association with treatment-related mortality and relapse. Blood 2002;100: 406–14. 5. Socié G, Stone JV, Wingard JR, et al. Long-term survival and late deaths after allogeneic bone marrow transplantation. Late Effects Working Committee of the International Bone Marrow Transplant Registry. N Engl J Med 1999; 341:14–21. 6. Antin JH, Ferrara JL. Cytokine dysregulation and acute graft-versus-host disease. Blood 1992;80:2964–8. 7. Ho VT, Soiffer RJ. The history and future of T-cell depletion as graft-versushost disease prophylaxis for allogeneic hematopoietic stem cell transplantation. Blood 2001;98:3192–204. 8. Urbano-Ispizua A, Solano C, Brunet S, et al. Allogeneic transplantation of selected CD34+ cells from peripheral blood: experience of 62 cases using immunoadsorption or immunomagnetic technique. Spanish Group of AlloPBT. Bone Marrow Transplant 1998;22:519–25. 9. Champlin RE, Passweg JR, Zhang MJ, et al. T-cell depletion of bone marrow transplants for leukemia from donors other than HLA-identical siblings: advantage of T-cell antibodies with narrow specificities. Blood 2000;95:3996– 4003. 10. Wagner JE, Thompson JS, Carter SL, Kernan NA. Effect of graft-versus-host disease prophylaxis on 3-year disease-free survival in recipients of unrelated donor bone marrow (T-cell Depletion Trial): a multi-centre, randomised phase II-III trial. Lancet 2005;366:733–41. 11. Pavletic SZ, Carter SL, Kernan NA, et al. Influence of T-cell depletion on chronic graft-versus-host disease: results of a multicenter randomized trial in unrelated marrow donor transplantation. Blood 2005;106:3308–13. 12. Bacigalupo A, Lamparelli T, Barisione G, et al. Thymoglobulin prevents chronic graft-versus-host disease, chronic lung dysfunction, and late transplantrelated mortality: long-term follow-up of a randomized trial in patients undergoing unrelated donor transplantation. Biol Blood Marrow Transplant 2006; 12:560–5. 13. Storb R, Deeg HJ, Whitehead J, et al. Methotrexate and cyclosporine compared with cyclosporine alone for prophylaxis of acute graft versus host disease after marrow transplantation for leukemia. N Engl J Med 1986;314:729–35. 14. Ratanatharathorn V, Nash RA, Przepiorka D, et al. Phase III study comparing methotrexate and tacrolimus (prograf, FK506) with methotrexate and cyclosporine for graft-versus-host disease prophylaxis after HLA-identical sibling bone marrow transplantation. Blood 1998;92:2303–14. 15. Nash RA, Antin JH, Karanes C, et al. Phase 3 study comparing methotrexate and tacrolimus with methotrexate and cyclosporine for prophylaxis of acute graft-versus-host disease after marrow transplantation from unrelated donors. Blood 2000;96:2062–8. 16. Cutler C, Li S, Ho VT, et al. Extended follow-up of methotrexate-free immunosuppression using sirolimus and tacrolimus in related and unrelated donor peripheral blood stem cell transplantation. Blood 2007;109:3108–14. 17. Bolwell B, Sobecks R, Pohlman B, et al. A prospective randomized trial comparing cyclosporine and short course methotrexate with cyclosporine and mycophenolate mofetil for GVHD prophylaxis in myeloablative allogeneic bone marrow transplantation. Bone Marrow Transplant 2004;34:621–5. 18. Martin PJ, Schoch G, Fisher L, et al. A retrospective analysis of therapy for acute graft-versus-host disease: initial treatment. Blood 1990;76:1464–72. 19. Mielcarek M, Storb R. Graft-vs-host disease after non-myeloablative hematopoietic cell transplantation. Leuk Lymphoma 2005;46:1251–60. 20. Przepiorka D, Smith TL, Folloder J, et al. Risk factors for acute graft-versushost disease after allogeneic blood stem cell transplantation. Blood 1999;94: 1465–70. 21. Glucksberg H, Storb R, Fefer A, et al. Clinical manifestations of graft-versushost disease in human recipients of marrow from HL-A-matched sibling donors. Transplantation 1974;18:295–304. 22. Przepiorka D, Weisdorf D, Martin P, et al. 1994 Consensus Conference on Acute GVHD Grading. Bone Marrow Transplant 1995;15:825–8. 23. Rowlings PA, Przepiorka D, Klein JP, et al. IBMTR Severity Index for grading acute graft-versus-host disease: retrospective comparison with Glucksberg grade. Br J Haematol 1997;97:855–64. 24. Van Lint MT, Uderzo C, Locasciulli A, et al. Early treatment of acute graftversus-host disease with high- or low-dose 6-methylprednisolone: a multicenter randomized trial from the Italian Group for Bone Marrow Transplantation. Blood 1998;92:2288–93. 25. Weisdorf D, Haake R, Blazar B, et al. Treatment of moderate/severe acute graft-versus-host disease after allogeneic bone marrow transplantation: an analysis of clinical risk features and outcome. Blood 1990;75:1024–30. 26. Roy J, McGlave PB, Filipovich AH, et al. Acute graft-versus-host disease following unrelated donor marrow transplantation: failure of conventional therapy. Bone Marrow Transplant 1992;10:77–82. 27. Cragg L, Blazar BR, Defor T, et al. A randomized trial comparing prednisone with antithymocyte globulin/prednisone as an initial systemic therapy for moderately severe acute graft-versus-host disease. Biol Blood Marrow Transplant 2000;6:441–7. 28. Lee SJ, Zahrieh D, Agura E, et al. Effect of up-front daclizumab when combined with steroids for the treatment of acute graft-versus-host disease: results of a randomized trial. Blood 2004;104:1559–64. 29. Carpenter PA, Sanders JE. Steroid-refractory graft-vs.-host disease: past, present and future. Pediatr Transplant 2003;7 Suppl 3:19–31. 30. Bianco JA, Higano C, Singer J, et al. The somatostatin analog octreotide in the management of the secretory diarrhea of the acute intestinal graft-versushost disease in a patient after bone marrow transplantation. Transplantation 1990;49:1194–5. 31. Ippoliti C, Champlin R, Bugazia N, et al. Use of octreotide in the symptomatic management of diarrhea induced by graft-versus-host disease in patients with hematologic malignancies. J Clin Oncol 1997;15:3350–4. 32. Miklos DB, Kim HT, Miller KH, et al. Antibody responses to H-Y minor histocompatibility antigens correlate with chronic graft-versus-host disease and disease remission. Blood 2005;105:2973–8. 33. Filipovich AH, Weisdorf D, Pavletic S, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graftversus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant 2005;11:945–56. 34. Shulman HM, Sullivan KM, Weiden PL, et al. Chronic graft-versus-host syndrome in man. A long-term clinicopathologic study of 20 Seattle patients. Am J Med 1980;69:204–17. 35. Koc S, Leisenring W, Flowers ME, et al. Therapy for chronic graft-versus-host disease: a randomized trial comparing cyclosporine plus prednisone versus prednisone alone. Blood 2002;100:48–51. 36. Arora M, Wagner JE, Davies SM, et al. Randomized clinical trial of thalidomide, cyclosporine, and prednisone versus cyclosporine and prednisone as initial therapy for chronic graft-versus-host disease. Biol Blood Marrow Transplant 2001;7:265–73. 37. Cutler C, Antin JH. Chronic graft-versus-host disease. Curr Opin Oncol 2006; 18:126–31. 38. Vogelsang GB. How I treat chronic graft-versus-host disease. Blood 2001;97: 1196–201. 39. Couriel D, Carpenter PA, Cutler C, et al. Ancillary therapy and supportive care of chronic graft-versus-host disease: national institutes of health consensus development project on criteria for clinical trials in chronic Graft-versus-host disease: V. Ancillary Therapy and Supportive Care Working Group Report. Biol Blood Marrow Transplant 2006;12:375–96. 40. Akpek G, Lee SJ, Flowers ME, et al. Performance of a new clinical grading system for chronic graft-versus-host disease: a multicenter study. Blood 2003; 102:802–9. test yourself with board-type questions Questions for self-assessment in selected specialties are available on Hospital Physician’s Web site. Go to www.turner-white.com, click on Hospital Physician, then click on “Board-Type Questions.” Copyright 2009 by Turner White Communications Inc., Wayne, PA. All rights reserved. 12 Hospital Physician Board Review Manual www.turner-white.com