Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Drug interaction wikipedia , lookup
Epinephrine autoinjector wikipedia , lookup
Compounding wikipedia , lookup
Prescription costs wikipedia , lookup
Pharmacokinetics wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Prescription drug prices in the United States wikipedia , lookup
Theralizumab wikipedia , lookup
Pharmaceutical industry wikipedia , lookup
Drug discovery wikipedia , lookup
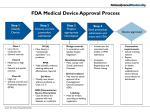
Premarket Testing and Validation Charles Lankford PharmaSys, Inc. 216 Towne Village Drive Cary, NC. 27513 [email protected] PharmaSys, Inc. www.pharma-sys.com 1 U S. Food and Drug Administration • Scientific, regulatory, and public health agency – oversees items accounting for 25 cents of every dollar spent by consumers. PharmaSys, Inc. • Jurisdiction – most food products (other than meat and poultry) – human and animal drugs – therapeutic agents of biological origin – medical devices – radiation-emitting products for consumer, medical, and occupational use – cosmetics – animal feed www.pharma-sys.com 2 History of the FDA • single chemist U.S. Department of Agriculture in 1862 • Regulatory functions added in1906 • The Bureau of Chemistry's name changed to the Food, Drug, and Insecticide Administration in July 1927, – nonregulatory research functions of the bureau were transferred elsewhere PharmaSys, Inc. www.pharma-sys.com 3 Events Leading to Formation of FDA 1905 - Samuel Hopkins Adams :11 articles The Great American Fraud in Collier's Weekly. – analyzed the contents of some of the country's most popular medicines. – argued that many of the companies producing these medicines were making false claims about their products. – some cases, these medicines were actually damaging the health of those people using them 1906 - Upton Sinclair: The Jungle – fictional portrait of life and death of working class immigrates in turn-ofthe-century Chicago – filled with page after page of nauseating detail he had researched about the meat-packing industry Public demanded sweeping reforms in the food industry 30 June 1906 President Roosevelt signed the Food and Drugs Act, known simply as the Wiley Act. PharmaSys, Inc. www.pharma-sys.com 4 1906 Food and Drugs Act • Prohibited interstate transport of unlawful food and drugs under penalty of seizure of the questionable products and/or prosecution of the responsible parties • Basis on regulation of product labeling rather than premarket approval • Drugs, defined in accordance with the standards of strength, quality, and purity in the United States Pharmacopoeia and the National Formulary, could not be sold in any other condition unless the specific variations from the applicable standards were plainly stated on the label PharmaSys, Inc. www.pharma-sys.com 5 1906 Food and Drugs Act • Foods were not defined according to analogous standards, but the law prohibited the addition of any ingredients that would substitute for the food, conceal damage, pose a health hazard, or constitute a filthy or decomposed substance • If the manufacturer opted to list the weight or measure of a food, this had to be done accurately • The food or drug label could not be false or misleading in any particular, and the presence and amount of eleven dangerous ingredients, including alcohol, heroin, and cocaine, had to be listed PharmaSys, Inc. www.pharma-sys.com 6 1911 • Supreme Court ruled that the law did not-apply to false therapeutic claims. PharmaSys, Inc. www.pharma-sys.com 7 The 1938 Food, Drug, and Cosmetic Act • A Tennessee drug company marketed a form of the new sulfa wonder drug, Sulfanilamide, a drug used to treat streptococcal infections, that would appeal to pediatric patients, Elixir Sulfanilamide. • 100 people in 15 states died, many were children • Dissolved in diethylene glycol, a highly toxic chemical analogue of antifreeze • Not tested for toxicity… food and drugs law did not require safety studies for new drugs PharmaSys, Inc. www.pharma-sys.com 8 The 1938 Food, Drug, and Cosmetic Act • legally mandated quality and identity standards for foods • prohibition of false therapeutic claims for drugs • coverage of cosmetics and medical devices • clarification of the FDA's right to conduct factory inspections • control of product advertising, among other items PharmaSys, Inc. www.pharma-sys.com 9 Kefauver-Harris Amendments • Thalidomide, chiefly sold and prescribed during the late 1950s and 1960s to pregnant women, as an antiemetic to combat morning sickness and as an aid to help them sleep • Drug stunted the growth of fetal arms and legs, 10,000 babies affected, never approved in US • Now approved for the treatment of MULTIPLE MYELOMA and ERYTHEMA NODOSUM LEPROSUM • Resulted in Kefauver-Harris Amendments PharmaSys, Inc. www.pharma-sys.com 10 Kefauver-Harris Amendments • mandates efficacy & safety before a drug could be marketed • requires FDA to assess the efficacy of all drugs introduced since 1938, • institutes stricter agency control over drug trials (including a requirement that patients involved must give their informed consent) • transferred from the Federal Trade Commission to the FDA regulation of prescription drug advertising, • established good manufacturing practices by the drug industry • granted the FDA greater powers to access company production and control records to verify those practices PharmaSys, Inc. www.pharma-sys.com 11 Drug or Device Deemed Adulterated if • If it consists in whole or in part of any filthy, putrid, or decomposed substance; PharmaSys, Inc. www.pharma-sys.com 12 Drug or Device Deemed Adulterated if • prepared, packed, or held under insanitary conditions • do not conform to or are not operated or administered in conformity with current good manufacturing practice • are not operated or administered in conformity with the positron emission tomography compounding standards and the official monographs of the United States Pharmacopoeia PharmaSys, Inc. www.pharma-sys.com 13 Drug or Device Deemed Adulterated if • composed, in whole or in part, of any poisonous or deleterious substance • a color additive which is unsafe • animal feed bearing or containing a new animal drug, and such animal feed is unsafe PharmaSys, Inc. www.pharma-sys.com 14 Drug or Device Deemed Adulterated if • strength differs from, or its quality or purity falls below, the standards • strength, quality, or purity shall be made in accordance with the tests or methods of assay set forth in such compendium PharmaSys, Inc. www.pharma-sys.com 15 Drug or Device Deemed Adulterated if • mixed or packed therewith so as to reduce its quality or strength or • substituted wholly or in part PharmaSys, Inc. www.pharma-sys.com 16 PART 211 – DRUG GMP • A--General Provisions • B--Organization and Personnel • C--Buildings and Facilities • D--Equipment • E--Control of Components and Drug Product Containers and Closures • F--Production and Process Controls PharmaSys, Inc. • G--Packaging and Labeling Control • H--Holding and Distribution • I--Laboratory Controls • Subpart J--Records and Reports • K--Returned and Salvaged Drug Products www.pharma-sys.com 17 Part 820 – Device QSR • A--General Provisions • B--Quality System Requirements • C--Design Controls • D--Document Controls • E--Purchasing Controls PharmaSys, Inc. • F--Identification and Traceability • G--Production and Process Controls • H--Acceptance Activities www.pharma-sys.com 18 Part 820 – Device QSR • I--Nonconforming Product • J--Corrective and Preventive Action • • K--Labeling and Packaging Control • K--Labeling and Packaging Control • N—Servicing • O--Statistical Techniques PharmaSys, Inc. L--Handling, Storage, Distribution, and Installation • M—Records www.pharma-sys.com 19 FDA Documentation • Regulation – Codified and Law • Preamble – Dialog of FDA’s thinking when reg was codified • Guidance - FDA’s current thinking about reg PharmaSys, Inc. www.pharma-sys.com 20 GCP and Clinical Trials • Electronic Records; Electronic Signatures (21 CFR Part 11) • Human Subject Protection (Informed Consent) (21 CFR Part 50) • Additional Safeguards for Children in Clinical Investigations of FDA-Regulated Products (Interim Rule) (21 CFR Part 50, subpart D) • Financial Disclosure by Clinical Investigators (21 CFR Part 54) • Institutional Review Boards (21 CFR Part 56) PharmaSys, Inc. www.pharma-sys.com 21 GCP and Clinical Trials • Forms 1571 (Investigational New Drug Application) and 1572 (Statement of Investigator) • Applications for FDA Approval to Market a New Drug (21 CFR Part 314) • Applications for FDA Approval of a Biologic License (21 CFR Part 601) • Investigational Device Exemptions (21 CFR Part 812) • Premarket Approval of Medical Devices (21 CFR Part 814 PharmaSys, Inc. www.pharma-sys.com 22 Drug Approval Process PharmaSys, Inc. www.pharma-sys.com 23 Drug Development and Approval Process • U.S. system most rigorous in the world • On average, it costs a company $360 million to get one new medicine from the laboratory to the pharmacist's shelf • It takes 12 years on average for an experimental drug to travel from lab to medicine chest. • Only five in 5,000 compounds that enter preclinical testing make it to human testing. • One of these five tested in people is approved. PharmaSys, Inc. www.pharma-sys.com 24 Biological Screening and Pharmacological Testing • Studies to explore the pharmacological activity and therapeutic potential of compounds. – animals, isolated cell cultures and tissues, enzymes and cloned receptor sites as well as computer models. • PharmaSys, Inc. www.pharma-sys.com 25 Pharmaceutical Dosage Formulation and Stability Testing • The process of turning an active compound into a form and strength suitable for human use . PharmaSys, Inc. www.pharma-sys.com 26 Toxicology and Safety Testing • Tests to determine the potential risk a compound poses to man and the environment – These studies involve the use of animals, tissue cultures, and other test systems to examine the relationship between factors such as dose level, frequency of administration, and duration of exposure to both the short- and long-term survival of living organisms. – LD50 PharmaSys, Inc. www.pharma-sys.com 27 Regulatory Review: Investigational New Drug (IND) Application • An application filed with the U.S. FDA prior to human testing. • The IND application is a compilation of all known information about the compound. It also includes a description of the clinical research plan for the product and the specific protocol for phase I study. • Unless the FDA says no, the IND is automatically approved after 30 days and clinical tests can begin. PharmaSys, Inc. www.pharma-sys.com 28 Phase I Clinical Evaluation • The first testing of a new compound in human subjects, for the purpose of establishing the tolerance of healthy human subjects at different doses • defining its pharmacologic effects at anticipated therapeutic levels • studying its absorption, distribution, metabolism, and excretion patterns in humans PharmaSys, Inc. www.pharma-sys.com 29 Phase II Clinical Evaluation • Controlled clinical trials of a compound's potential usefulness and short term risks. • A relatively small number of patients, usually no more than several hundred subjects, enrolled in phase II studies. PharmaSys, Inc. www.pharma-sys.com 30 Phase III Clinical Evaluation • Controlled and uncontrolled clinical trials of a drug's safety and effectiveness in hospital and outpatient settings. – gather precise information on the drug's effectiveness – determine adverse effects – identify the best way of administering and using – phase III studies can involve several hundred to several thousand subjects. PharmaSys, Inc. www.pharma-sys.com 31 Institutional Review Boards • • • • • • used to ensure the rights and welfare of people participating in clinical trials both before and during their trial participation. make sure that participants are fully informed and have given their written consent before studies ever begin. monitored by the FDA to protect and ensure the safety of participants in medical research. An IRB must be composed of no less than five experts and lay people with varying backgrounds to ensure a complete and adequate review of activities commonly conducted by research institutions. In addition to possessing the professional competence needed to review specific activities, an IRB must be able to ascertain the acceptability of applications and proposals in terms of institutional commitments and regulations, applicable law, standards of professional conduct and practice, and community attitudes. Therefore, IRBs must be composed of people whose concerns are in relevant areas. PharmaSys, Inc. www.pharma-sys.com 32 Bioavailability Studies • The use of healthy volunteers to document the rate of absorption and excretion from the body of a compound's active ingredients. • Companies conduct bioavailability studies both at the beginning of human testing and just prior to marketing to show that the formulation used to demonstrate safety and efficacy in clinical trials is equivalent to the product that will be distributed for sale. • Companies also conduct bioavailability studies on marketed products whenever they change the method used to administer the drug (e.g., from injection or oral dose form), the composition of the drug, the concentration of the active ingredient, or the manufacturing process used to produce the drug. PharmaSys, Inc. www.pharma-sys.com 33 Process Development for Manufacturing and Quality Control (CMC) • Engineering and manufacturing design activities to establish a company's capacity to produce a product in large volume and development of procedures to ensure chemical stability, batch-to-batch uniformity, and overall product quality. PharmaSys, Inc. www.pharma-sys.com 34 Regulatory Review: New Drug Application (NDA) • An application to the FDA for approval to market a new drug. All information about the drug gathered during the drug discovery and development process is assembled in the NDA. • During the review period, the FDA may ask the company for additional information about the product or seek clarification of the data contained in the application. PharmaSys, Inc. www.pharma-sys.com 35 Postapproval Research • Adverse events must be reported • Additional indications PharmaSys, Inc. www.pharma-sys.com 36 Basic Regulatory Requirements for Medical Devices • Premarket Notification 510(k), unless exempt, or Premarket Approval (PMA) • Establishment registration on form FDA2891 • Medical Device Listing on form FDA-2892 • Quality System (QS) regulation • Labeling requirements • Medical Device Reporting (MDR) PharmaSys, Inc. www.pharma-sys.com 37 Premarket Notification 510(k) 21 CFR Part 807 Subpart E • If device requires the submission, – can not commercially distribute until you receive a letter of substantial equivalence from FDA authorizing you to do so. PharmaSys, Inc. www.pharma-sys.com 38 Premarket Notification 510(k) 21 CFR Part 807 Subpart E • A 510(k) must demonstrate that the device is substantially equivalent to one legally in commercial distribution – Application fee applies to Traditional, Abbreviated, and Special 510(k)s. Small businesses may pay smaller fee. PharmaSys, Inc. www.pharma-sys.com 39 Premarket Notification 510(k) 21 CFR Part 807 Subpart E • Most Class I devices and some Class II devices are exempt – A list of exempt devices is located at: http://www.accessdata.fda.gov/scripts/cdrh/cf docs/cfpcd/315.cfm PharmaSys, Inc. www.pharma-sys.com 40 Premarket Approval (PMA) - 21 CFR Part 814 • Product requiring PMAs – Class III devices, high risk devices, pose a significant risk of illness or injury – devices found not substantially equivalent to Class I and II predicate PharmaSys, Inc. www.pharma-sys.com 41 Premarket Approval (PMA) - 21 CFR Part 814 • PMA process is more involved – includes the submission of clinical data to support claims made for the device. – The PMA is an actual approval of the device by FDA. • Medical device user fees apply to original PMAs and certain types of PMA supplements. PharmaSys, Inc. www.pharma-sys.com 42 Investigational Device Exemption (IDE) - 21CFR Part 812 • Clinical studies with devices of significant risk must be approved by FDA and by an Institutional Review Board (IRB) before the study can begin. • Studies with devices of nonsignificant risk must be approved by the IRB only PharmaSys, Inc. www.pharma-sys.com 43 Establishment Registration form FDA-2891 - 21 CFR Part 807 • Manufacturers must register their establishments with the FDA • Once a year, FDA sends the registration form FDA-2891(a) to all registered firms to be verified, corrected, and returned by the firm as a yearly registration • foreign manufacturers must also designate a U.S. Agent PharmaSys, Inc. www.pharma-sys.com 44 Medical Device Listing form FDA-2892 - 21CFR Part 807 • All medical devices are required to be listed with the • Firms that are required to list their devices are those that: – – – – – – manufacture, repackage and relabel, develop specifications, reprocess single-use devices, remanufacture manufacture accessories and components sold directly to the end user PharmaSys, Inc. www.pharma-sys.com 45 Labeling - 21 CFR Part 801 • Labeling includes labels on the device as well as descriptive and informational literature that accompanies the device. Labeling requirements can be accessed on the web at: http://www.fda.gov/cdrh/devadvice/33.html PharmaSys, Inc. www.pharma-sys.com 46 Medical Device Reporting - 21 CFR Part 803 • Incidents in which a device may have caused or contributed to a death or serious injury must to be reported to FDA under the Medical Device Reporting program. • The MDR regulation is a mechanism for FDA and manufacturers to identify and monitor significant adverse events involving medical devices. The goals of the regulation are to detect and correct problems in a timely manner. PharmaSys, Inc. www.pharma-sys.com 47 Medical Device Classification • classifications for approximately 1,700 different generic types of devices • three regulatory classes based on the level of control necessary to assure the safety and effectiveness of the device. PharmaSys, Inc. www.pharma-sys.com 48 Device Class and Regulatory Controls • Class I General Controls – With Exemptions – Without Exemptions • Class II General Controls and Special Controls – With Exemptions – Without Exemptions • Class III General Controls and Premarket Approval PharmaSys, Inc. www.pharma-sys.com 49 Class I Devices • Class I devices are subject to the least regulatory control. • They present minimal potential for harm to the user and are often simpler in design than Class II or Class III devices. • Most Class I devices are exempt from the premarket notification and/or good manufacturing practices regulation. Information on Class I exempt devices is located under the heading What are Class I/II Exemptions?. PharmaSys, Inc. www.pharma-sys.com 50 Class I - General Controls • Class I devices are subject to "General Controls” – – – – Establishment Registration ( Medical Device Listing Manufactured s in accordance with GMP Labeled in accordance with labeling regulations (21 CFR Part 801 or 809) – Submission of a premarket notification [510(k)] Examples of Class I devices include elastic bandages, examination gloves, and hand-held surgical instruments. PharmaSys, Inc. www.pharma-sys.com 51 Class II - Special Controls • Special controls may include special labeling requirements, mandatory performance standards and postmarket surveillance. Examples of Class II devices include sterilizers, LIMS, powered wheelchairs, infusion pumps, and surgical drapes. PharmaSys, Inc. www.pharma-sys.com 52 Class III - Premarket Approval • Class III is the most stringent regulatory category for devices. • insufficient information exists to assure safety and effectiveness • Class III devices are usually those that support or sustain human life • Premarket approval is required • Not all Class III devices require an approved PMA. – Class III devices which are equivalent to devices legally marketed before May 28, 1976 PharmaSys, Inc. www.pharma-sys.com 53 Class III Devices Requiring an Approved Premarket Approval Application Examples of Class III devices which require a premarket approval include replacement heart valves, silicone gel-filled breast implants, and implanted cerebella stimulators, pacemaker. PharmaSys, Inc. www.pharma-sys.com 54 DESIGN VERIFICATION AND VALIDATION PharmaSys, Inc. www.pharma-sys.com 55 DESIGN VERIFICATION AND VALIDATION • Establish and maintain procedures for verifying device design • Confirm design output meets the design input requirements • Results documented in the DHF PharmaSys, Inc. www.pharma-sys.com 56 DESIGN VERIFICATION AND VALIDATION • Performed under defined operating conditions on initial production units, lots, or batches, or their equivalents • Include testing of production units under actual or simulated use conditions • Include software validation and risk analysis, where appropriate PharmaSys, Inc. www.pharma-sys.com 57 DEFINITIONS • Specification - any requirement with which a product, process, service, or other activity must conform • Validation - confirmation by examination and provision of objective evidence that the particular requirements for a specific intended use can be consistently fulfilled PharmaSys, Inc. www.pharma-sys.com 58 DEFINITIONS • Process Validation - objective evidence that a process consistently produces a result or product meeting specifications • Design Validation - objective evidence that device specifications conform with user needs and intended use(s) • Verification - confirmation by examination and provision of objective evidence that specified requirements have been fulfilled PharmaSys, Inc. www.pharma-sys.com 59 DESIGN VERIFICATION AND VALIDATION • Always done versus specifications • Control of specifications increases probability of achieving desired results • Specifications reviewed before as development procedes. • As designs evolve, they should be evaluated versus their current specifications. PharmaSys, Inc. www.pharma-sys.com 60 DESIGN VERIFICATION AND VALIDATION • Always done versus specifications • Control of specifications increases probability of achieving desired results • Specifications reviewed before as development procedes. • As designs evolve, they should be evaluated versus their current specifications. PharmaSys, Inc. www.pharma-sys.com 61 DESIGN VERIFICATION AND VALIDATION • Should be done with test equipment calibrated and controlled according to quality system requirements. PharmaSys, Inc. www.pharma-sys.com 62 DESIGN VERIFICATION AND VALIDATION • Performed according to a written protocol(s) • Include defined conditions for the testing • Approved before being used. PharmaSys, Inc. www.pharma-sys.com 63 DESIGN VERIFICATION AND VALIDATION • Test protocol(s) are not perfect … • annotate ongoing changes to a protocol • record technical comments about any deviations or other events that occurred during testing • The slightest problem should not be ignored PharmaSys, Inc. www.pharma-sys.com 64 SOFTWARE VALIDATION • Ongoing development - evaluated and reviewed versus the software specifications • “Final" prototype(s) - software and hardware are validated PharmaSys, Inc. www.pharma-sys.com 65 SOFTWARE VALIDATION • Before testing detailed code should be visually reviewed versus flow charts and specifications. • All cases, especially decision points and error/limit handling, should be reviewed and the results documented. PharmaSys, Inc. www.pharma-sys.com 66 SOFTWARE VALIDATION • Code visually reviewed versus flow charts and specifications • All cases… decision points and error/limit handling, reviewed and the results documented PharmaSys, Inc. www.pharma-sys.com 67 SOFTWARE VALIDATION • Algorithms should checked for accuracy. – Recalls have occurred because algorithms were incorrectly copied from a source and, in other cases, because the source algorithm was incorrect. PharmaSys, Inc. www.pharma-sys.com 68 SOFTWARE VALIDATION • Testing includes normal operation of the complete device • Combined system of hardware and software should be challenged with abnormal inputs and conditions. As appropriate, these inputs and conditions include such items as PharmaSys, Inc. www.pharma-sys.com 69 Inputs and Conditions • operator errors • induced failure of sensors and cables or other interconnects • induced failure of output equipment PharmaSys, Inc. • exposure to static electricity • power loss and restart • simultaneous inputs or interrupts • limit testing www.pharma-sys.com 70 SOFTWARE VALIDATION • Testing includes normal operation of the complete device • Combined system of hardware and software should be challenged with abnormal inputs and conditions. As appropriate, these inputs and conditions include such items as PharmaSys, Inc. www.pharma-sys.com 71 LABELING VERIFICATION • Exercised such that all labeling, displays, and outputs are generated, reviewed, and the results documented. • All displayed prompts and instructions are checked versus the manufacturer's and FDA's labeling requirements and operator manual. PharmaSys, Inc. www.pharma-sys.com 72 LABELING VERIFICATION • Printed labeling and screen displays should be checked to see if they are directed to the user and not to the system designer • Data, identifications, or other key information displayed should be current, complete, unambiguous, and accurate PharmaSys, Inc. www.pharma-sys.com 73 LABELING VERIFICATION • Printouts should undergo a verification similar to that performed for the screen or other displays • Annotated copies of verified labeling, printouts, etc. and associated notes and any checklists should be placed in the design history file PharmaSys, Inc. www.pharma-sys.com 74 § 820.30(h) Design Transfer • Each manufacturer shall establish and maintain procedures to ensure that the device design is correctly translated into production specifications. – Production specifications must ensure that manufactured devices are repeatedly and reliably produced within product and process capabilities. – The process of encapsulating knowledge about the device into production specifications is critical to device quality. PharmaSys, Inc. www.pharma-sys.com 75 § 820.30(j) Design history file • Each manufacturer shall establish and maintain a DHF for each type of device. – Other national regulations require some form of documentation and records. Product documentation required by Canada, Europe, and Japan contain certain elements of the U. S. FDA design history file requirements without requiring all the elements to be compiled in a file. – Virtually every section of the design control requirements specifies information which should be recorded. The compilation of these records is sometimes referred to as the design history file. PharmaSys, Inc. www.pharma-sys.com 76 Typical DHF • Detailed design and development plan specifying design tasks and deliverables. • Copies of approved design input documents and design output documents. • Documentation of design reviews. • Validation documentation. • When applicable, copies of controlled design documents and change control records. PharmaSys, Inc. www.pharma-sys.com 77 Design changes • establish and maintain procedures for design changes before their implementation. – Document control – Change control PharmaSys, Inc. www.pharma-sys.com 78 Production Testing is not Verification • Some manufacturers erroneously equate production testing with verification. – Verification testing establishes conformance of design output with design input – Production testing is to determine whether the unit under test has been correctly manufactured… screen out manufacturing process errors and detect infant mortality failures • Typically, a small subset of functional and performance tests accomplish this objective with a high degree of accuracy. • Production testing is rarely, if ever, comprehensive enough to verify the design. PharmaSys, Inc. www.pharma-sys.com 79 Example A leakage test may be used during production to ensure that a hermetically-sealed enclosure was properly assembled … may not be sensitive enough to detect long-term diffusion of gas through the packaging material. Permeability of the packaging material is an intrinsic property of the material rather than an assembly issue… likely be verified using a more specialized test than is used during production. PharmaSys, Inc. www.pharma-sys.com 80 Environmental Conditions • Validation should include simulation of the expected environmental conditions PharmaSys, Inc. www.pharma-sys.com 81 VALIDATION DOCUMENTATION • Validation is a compilation of the results of all validation activities. • For a complex design, the detailed results may be contained in a variety of separate documents and summarized in a validation report. • Supporting test articles should be explicitly referenced in the validation report and either included as an appendix or available in the design history file. PharmaSys, Inc. www.pharma-sys.com 82 WHAT PROCESSES SHOULD BE VALIDATED • Routine end-product tests have insufficient sensitivity to verify the desired safety and efficacy of the finished devices • Clinical or destructive testing would be required to show that the manufacturing process has produced the desired result or product • Routine end-product tests do not reveal all variations in safety and efficacy that may occur in the finished devices. • The process capability is unknown, or it is suspected that the process is barely capable of meeting the device specifications. PharmaSys, Inc. www.pharma-sys.com 83 § 820.75 PROCESS VALIDATION • (a) Where the results of a process cannot be fully verified by subsequent inspection and test, the process shall be validated with a high degree of assurance and approved according to established procedures. – The validation activities and results, including the date and signature of the individual(s) approving the validation and where appropriate the major equipment validated, shall be documented. • (b) Each manufacturer shall establish and maintain procedures for monitoring and control of process parameters for validated processes to ensure that the specified requirements continue to be met. – (1) Each manufacturer shall ensure that validated processes are performed by qualified individual(s). – (2) For validated processes, the monitoring and control methods and data, the date performed, and, where appropriate, the individual(s) performing the process or the major equipment used shall be documented. • (c) When changes or process deviations occur, the manufacturer shall review and evaluate the process and perform revalidation where appropriate. These activities shall be documented. PharmaSys, Inc. www.pharma-sys.com 84 Planning the Process Validation Study • The plan should include design reviews. • The plan for the validation study is documented in the validation protocol. • A copy of the protocol and validation results are placed in the Design History File (DHF) [820.30 (j)] or quality system record file (820.186). • The operational, monitoring, and other production-related procedures are part of the device master record (DMR) (820.181). PharmaSys, Inc. www.pharma-sys.com 85 Typical Elements of Process Validation Study • • • • • • • • • • • identification of the process identification of device(s) to be manufactured using the process criteria for a successful study length and duration of the study assumptions (shifts, operators, equipment, components; identification of equipment to be used in the process [820.75(b)(2)] identification of utilities for the process equipment identification of operators and required operator qualifications [820.75(b)(2)]; complete description of the process {may reference the DMR [820.181(b)]}; relevant specifications including those for the product, components, manufacturing materials, the environment, etc. [may reference the DMR and quality system files {820.181(a) and (b); 820.186}; PharmaSys, Inc. • • • • • • • • • any special controls or conditions to be placed on preceding processes during the validation; process parameters to be controlled and monitored, and methods for controlling and monitoring [820.70(a); 820.75(b)(2)]; product characteristics to be monitored and method for monitoring [820.70(a)(2); 820.75(b)(2); 820.80(c)]; any subjective criteria used to evaluate the product; definition of what constitutes nonconformance for both measurable and subjective criteria; statistical methods for data collection and analysis (820.250); consideration of maintenance and repairs [820.72(a)]; conditions that may indicate that the process should be revalidated [820.75(c)]; stages of the study where design review is required; and approval(s) of the protocol. www.pharma-sys.com 86 Installation and Operation Qualification • Process equipment should be installed, reviewed, calibrated, challenged, and evaluated to ensure that it is capable of operating within established limits and tolerances as well as throughout all anticipated operating ranges. – – – – – – – – – – – examining equipment design and supplied documentation; determining installation requirements; establishing any needed environmental controls and procedures; assuring that the work area has sufficient space to perform the processing and associated activities; installing the equipment; verifying correct installation; establishing manufacturing procedures for the monitoring, operation, and control of the equipment including the minimum number of operators; determining calibration, cleaning, maintenance, adjustment, and expected repair requirements; identifying important elements of the equipment that could affect the output or finished device; verifying that the system or subsystem performs as intended throughout all anticipated operating ranges; and documenting the above information. PharmaSys, Inc. www.pharma-sys.com 87 Process Performance Qualification • The purpose of process performance qualification is to rigorously test the process to determine whether it is capable of consistently producing an output or inprocess or finished devices which meet specifications. In entering the process performance qualification phase of validation, it is understood that the: – device, packaging, and process specifications have been established, documented, and essentially proven acceptable through engineering, laboratory or other verification methods [820.30; 820.70(a)]; and – process and ancillary equipment and the environment have been judged acceptable on the basis of installation and operation qualification studies [820.70(g)]. PharmaSys, Inc. www.pharma-sys.com 88 Product Performance Qualification • Demonstrate that the process has not adversely affected the finished product and that the product meets its predetermined specifications and quality attributes. • Products used for design validation should be manufactured using the same production equipment, methods and procedures that will be used in routine production. • Design validation can be conducted using finished products made during process validation studies and will satisfy the need for product performance qualification. • Design validation shall ensure that devices conform to defined user needs and intended uses and shall include testing production units under actual or simulated use conditions [820.30(g)]. • Original designs and design changes are subject to design control requirements [820.30(i)]. The results of design validation are subject to review under the design control review requirements [820.30(e)]. PharmaSys, Inc. www.pharma-sys.com 89 Questions & Answers • Charles Lankford • PharmaSys, Inc. • 216 Towne Village Drive • Cary, NC. 27513 • [email protected] integrity… commitment… value… results… PharmaSys, Inc. www.pharma-sys.com 90