Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
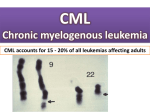
- Slower rate of cellular prolifiration - Sometimes discovered accidentaly during a routine bL count. -Cells are more mature ----> gain access to the circulation more easy ---> very high WBCs count. On the otherhand overcrowding of the marrow occurs late ----> weakness, bleeding tendency occur relatively late. -Thus this early presentation are the effect of metastasis e.g. splenomegaly which may be huge. Characterized by prolifiration of white cells which invade the blood stream, may infiltrate any part of the body. Definition:It is a form of leukemia characterized by the increased and unregulated growth of predominantly myeloid cells in the bone marrow and the accumulation of these cells in the blood. CML is a clonal bone marrow stem cell disorder in which proliferation of mature granulocytes (neutrophils, eosinophils, and basophils) and their precursors is the main finding.. Epidemiology: CML occurs in all age groups, but most commonly in the middle-aged and elderly. Its annual incidence is 1–2 per 100,000 people, and slightly more men than women are affected. CML represents about 15– 20% of all cases of adult leukemia in Western populations.The only well-described risk factor for CML is exposure to ionizing radiation; for example, increased rates of CML were seen in people exposed to the atomic bombings of Hiroshima and Nagasaki[16 It is a type of myeloproliferative disease associated with a characteristic chromosomal translocation called the Philadelphia chromosome CML was the first malignancy to be linked to a clear genetic abnormality, the chromosomal translocation known as the Philadelphia chromosome. This chromosomal abnormality is so named because it was first discovered and described in 1960 by two scientists from Philadelphia,Pennsylvania: This exchange brings together two genes: the BCR (breakpoint cluster region) gene on chromosome 22 and the proto-oncogene ABL (Ableson leukemia virus) on chromosome 9. The resulting hybrid gene BCR-ABL codes for a fusion protein with tyrosine kinase activity, which activates signal transduction pathways, leading to uncontrolled cell growth. The Philadelphia chromosome: T (9:22) translocation. The Ph chromosome is a shortened chromosome 22, which result from Reciprocal translocation between the long arms of chromosomes 9 & 22 ,The result is a hybrid bcr-abl gene (fusion protein) with increased tyrosine Kinase acm resulting in leukemic transformation. AGE: CML is a disease of young adults ( ) 20 - 45 years, but may occur at any age. Sex: Females more than males. Onset: Gradual. Course : Spontenous remissions and exacerbations. The disease generally terminates in marrow transformation (AML) or failure associated with fibrosis. Prognosis Patient usually die within 6 months --> 2 years but may live for 5 years or more. Cl. Picture : Symptoms: * may be diagnosed accidentaly. * general lassitude, rapid fatigability, bone ache. * Symptoms caused by splenomegally : --> dragging Lt hypochondrial pain, may be: ---> acute Lt hypochondrial pain. ---> abd. swelling ---> dyspepsia *Symptoms caused by leukemic infilterations: *Skin : Pruritis . nodules, eruptions, pigmentations *Chest : cough and hemoptysis. *urinary : hematuria. *GIT - Haemorrhage. -discomfort or pain from gastric infilterations *Genital: Priampism in males, infertility and haemorrhage in females *Eyes : impairment of vision (haemorrhage-exudate( *Symptoms 2 ry to thrombocytopenia :(bleeding gums, other bleeding tendency( *Symptoms 2 ry to acute exacerbations: - fever -sore throat - bone ache - Haemorrhage Signs: 1- General : -low grade fever if exacerbations --> high fever -pallor -under weight - Tender sternum. 2-Splenomegally (Huge, Firm, irregular, may be areas of tenderness (perisplenism), may be splenic rub (over areas of infarction) 3- Lymphadenopathy : uncommon 4- Hepatomegally : variable 5- Signs of leukemic infilterations Skin N.S Laboratory Findings: 1 -Blood picture : RBCs . very early may be polycythemia, late anemia. platelets: very early may be rise, late decrease WBCs : Total count: may be very high () 200 - 400.000 Differential count: predominantely myelocytes, some mature granuulocytes, Myeloblastes (1- 5%) if more --> acute exacerbation Basophils, Eosinophils are increased. The total count may drop if: - Spont. remission -Therapeutic remission -Acute exacerbation. -Acute pyogenic infection Leucocyte alkaline phosphatase: (LAP) score This test was once used to see if the cause of a high white blood cell count might be CML. It is rarely used now, since there are better ways to check the blood for suspected CML. The leucocyte alkaline phosphatase (LAP) score is a cytochemistry test -- cells from the sample are placed on microscope slides, and chemical stains (dyes) that react only with certain types of cells are added. The stains cause color changes which can be seen only under a microscope. Normally the LAP score goes up as the white blood cell (WBC) count goes up. But people with CML tend to have high WBC counts with low LAP scores Ultimately, CML is diagnosed by detecting the Philadelphia chromosome. This characteristic chromosomal abnormality can be detected by routine cytogenetics, by fluorescent in situ hybridization, or by PCR for the bcr-abl fusion gene 2 -BM Aspiration: complete replacement of fat by cellular elements mostly granulocytes, but few blasts with exclusion of other hemopatopoietic elements. Diff. Diagnosis: 1- leukemoid reaction: syndrome with morphologic changes similar to leukemia in peripheral blood usually 2ry to infections:T.B., pneumonia, meningitis or metabolic causes But * Eosinophils and bosophils are decreased (rather than increased). *WBCs are alkaline phosphatase stongly +ve (rather than -ve) *B.M. is only moderately hyperplastic. 2-Myelofibrosis : splenomegally, only moderate leukocytosis, Fibrotic marrow. WBCs ALK. phosph +ve, no philadephia chromosome Classification: CML is often divided into three phases based on clinical characteristics and laboratory findings. CML typically begins in the chronic phase, and over the course of several years progresses to an accelerated phase and ultimately to a blast crisis. Blast crisis is the terminal phase of CML and clinically behaves like an acute leukemia. One of the drivers of the progression from chronic phase through acceleration and blast crisis is the acquisition of new chromosomal abnormalities (in addition to the Philadelphia chromosome).Some patients may already be in the accelerated phase or blast crisis by the time they are diagnosed. Chronic phase: Approximately 85% of patients with CML are in the chronic phase at the time of diagnosis. During this phase, patients are usually asymptomatic or have only mild symptoms of fatigue or abdominal fullness. The duration of chronic phase is variable and depends on how early the disease was diagnosed as well as the therapies used. Ultimately, in the absence of curative treatment, the disease progresses to an accelerated phase. Accelerated phase Criteria for diagnosing transition into the accelerated phase are somewhat variable; the most widely used criteria are those put forward by investigators at M.D. The World Health Organization.The WHO criteria are perhaps most widely used, and define the accelerated phase by any of the following: *10–19% myeloblasts in the blood or bone marrow *>20% basophils in the blood or bone marrow *Platelet count <100,000, unrelated to therapy *Platelet count >1,000,000, unresponsive to therapy . *Cytogenetic evolution with new abnormalities in addition to the Philadelphia chromosome *Increasing splenomegaly or white blood cell count, unresponsive to therapy The patient is considered to be in the accelerated phase if any of the above are present. The accelerated phase is significant because it signals that the disease is progressing and transformation to blast crisis is imminent Blast crisis Blast crisis is the final phase in the evolution of CML, and behaves like an acute leukemia, with rapid progression and short survival. Blast crisis is diagnosed if any of the following are present in a patient with CML: *>20% myeloblasts or lymphoblasts in the blood or bone marrow *Large clusters of blasts in the bone marrow on biopsy *Development of a chloroma (solid focus of leukemia outside the bone marrow) Treatment Chronic phase Chronic phase CML is treated with inhibitors of tyrosine kinase, the first of which was imatinib mesylate (marketed as Gleevec or Glivec. In the past, antimetabolites (e.g. cytarabine, hydroxyurea), alkylating agents, interferon alfa 2b, and steroids were used, but these drugs have been replaced by imatinib. Imatinib was approved by the United States FDA in 2001 and specifically targets BCR/abl, the constitutively activated tyrosine kinase fusion protein caused by the Philadelphia chromosome translocation. Bone marrow transplantation was also used as initial treatment for CML in younger patients before the advent of imatinib, and while it can often be curative, there was a high rate of transplant-related mortality. The transplantrelated mortality rate in the present is less than 5%. A number of newer drugs are being used to treat the minority of patients who develop imatinib resistance. To overcome imatinib resistance and to increase responsiveness of TK inhibitors, two novel agents have been developed. The first, dasatinib, is a TK inhibitor that blocks several oncogenic proteins and has been approved by the US FDA to treat CML patients who are either resistant to or intolerant of imatinib in 2007. Another TK inhibitor, nilotinib, is also approved by the US FDA for the same indication Dasatanib and nilotinib failed to overcome the imatinib resistance caused by the T315I mutation. All current treatments for this mutation are experimental. Recently omacetaxine, administered subcutaneously in CML patients who had failed imatinib and who have the highly drug resistant T315I kinase domain mutation. Stem cell transplantation is an option for those patients who developed T315I mutation. In 2005 favourable results of vaccination were reported with the BCR/abl p210 fusion protein in patients with stable disease, with GM-CSF as an adjuvant Prognosis : Three different risk groups were identified based on a prognostic scoring system that includes several variables: age, spleen size, blast count, platelet count, eosinophil count and basophil count.. In the lowest risk group, the median survival time was 98 months. In the middle group, the median was 65 months, and in the highest risk group, the median was about 42 months. Of all patients analyzed, the longest survival time was 117 months. However, this study pre-dates the advent of treatments using targeted therapy. A follow-up on patients using imatinib published in the New England Journal of Medicine shows an overall survival rate of 89% after five years