Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
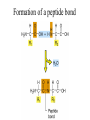
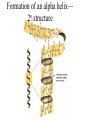
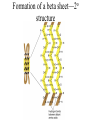
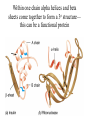
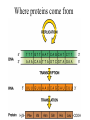
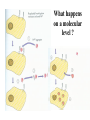
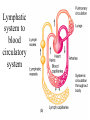
PRIONS Defn: small proteinaceous infectious particles that resist inactivation by procedures that modify viruses and nucleic acids Prion / Amyloid Diseases Prion Diseases •High levels of misfolded prion proteins •Transmissible Amyloid Diseases •Amyloid Fibers Found in Brain •Not Transmissible Scrapie BSE Kuru Creutzfeldt-Jakob disease Alzheimer’s (~ 4.5 million, US) Parkinson’s (~ 500,000, US) Huntington’s (~ 30,000 US) Prion /Amyloid diseases are usually neurodegenerative. Courtesy of Sid Taylor, MUW CJD is a neurological disease Occurs sporadically in humans at a ratio of 1 per 1 million people Estimated that 1 per 10,000 people have CJD at the time of death (estimate could be inaccurate as CJD could be mistaken for similar neurological diseases) Two types of CJD (genetic— hereditary and infectious) 1. CJD naturally occurring due to a mutation in a gene that encodes the PrPc neural protein. 2. vCJD infectious caused by consuming beef from cattle with BSE who are infected with the PrPsc protein The neural proteins PrPc and PrPsc look different so one can tell the difference between infectious and hereditary CJD PrPsc is only transmissible from one human to another via corneal transplants brain surgery with contaminated instruments contaminated brain probes CJD--Pathology Post-mortem examination of brain and neural tissue and cells show amyloid (starch-like) protein deposits—plaques between cells Non-inflammatory lesions Strange formations of neurons Vacuoles—large membrane bound inclusion bodies CJD—clinical manifestations Naturally occurring CJD affects humans at the age of 50-60 vCJD from transmissible prions can affect people as early as 14 Disease manifestations shaking loss of motor control Dementia—memory loss paralysis pneumonia death—a few months to 1.5 years after first symptoms Prion Properties • Prions are misfolded proteins. – Prion conformation is rich in b-sheets. • Prions aggregate in amyloids. • Prions are infectious. – Prion proteins induce conformational changes in other like proteins. • Prions can propagate into other cells. Courtesy of Sid Taylor/MUW What are proteins Proteins are made up of amino acids—each amino acid has its own Chemical properties The amino acids are linked together in by peptide bonds to form a Primary Sequence like “beads on a string”--polypeptide The primary sequence can form higher ordered structures called Secondary Structures—these can be alpha helices or beta sheets Region that has formed secondary structures can fold further to bring these secondary structures together to form a tertiary structure. Two or more identical or different tertiary structures come together to form quaternary structures (more than one subunit) Formation of a peptide bond Formation of an alpha helix— o 2 structure Formation of a beta sheet—2o structure Within one chain alpha helices and beta sheets come together to form a 3o structure— this can be a functional protein If more than one tertiary structure form interactions a 4o structure is formed Where proteins come from How proteins are made in eukaryotic cells Where do proteins end up? 1. Stay in the cytoplasm 2. Can be transported to the surface of the cell—membrane protein 3. Can be secreted from the cell and into the external milieu --they may stay near the cells that secreted them --they may be circulated throughout the body Summary of previous notes 1. CJD genetic anomaly vs. vCJD caused by consuming cows with BSE. 2. PrPc (Prion protein cellular), alpha helical makeup, associated with brain cell surface. Function unknown. 3. PrPsc (Prion protein that looks like protein that causes scrapies in sheep), beta sheet structure, not associated with the cell surface. Summary of previous notes CJD genetic anomaly in humans 1. PrPc is normally found on the surface of brain cells. 2. In some individuals a rare mutation on chromosome 20 can cause PrPc to misfold into PrPsc . 3. PrPsc dissociates from the cell membrane but causes PrPc that is associated with the cell membrane to misfold and dissociate from the cell surface. 4. As PrPsc dissociates from the cell surface, more PrPc is translated in brain cell and translocated to the surface/ PrPsc induces misfolding and release of these proteins. Etcetera. 5. The accumulation of cell free PrPsc forms proteinaceous plaques between the brain cells 6. Aggregated PrPsc is finally internalized into cells giving cells the spongiform appearance. What happens on a molecular level ? Summary of previous notes vCJD variant CJD from consuming infected beef from cows with BSE • BSE in cattle is analogous to CJD in humans in that it is a genetic anomaly that leads to misfolding cell associated prion proteins into pathological cell free proteins. 2. In later stages of the disease process this is manifested as Mad Cow Disease. 3. The PrPsc from the cow can be transmitted to humans when humans consume contaminated meat. 4. The misfolded protein enters the human’s nerve cells and travels to the brain tissue. 5. The pathological proteins from the cow causes the PrPc that is naturally associated with the human cell membrane to misfold and dissociate from the cell surface. Epidemiology PrPsc so far only transmissible to man from cows with BSE Cattle can also transmit disease to domestic cats, sheep and pigs Transmission from sheep and pigs to humans so far not documented CWD –chronic wasting disease in elk and muledeer--a disease similar to BSE in cattle not documented as transmissible to humans PrPsc—not transmissible through milk or milk products Probable cause Hereditary BSE occurs naturally in cows at a low rate as CJD does in humans. 1. Cows are butchered before they show manifestations of disease such that PrPsc enters the human population at a low rate 2. Can be passed to the offspring and found associated with placenta that contaminates grass that cows graze on 3. Farmers put ground up cattle in cattle feed, spreading BSE in the bovine community—PrPsc enters the human population at a high rate. PrPsc is spread from cows with BSE to humans to cause vCJD by consumption of such cows PrPsc has been found concentrated in the following areas in cows brain spinal cord retina (eye) distal ileum (small intestines) neurons near the backbone bone marrow lymphatic tissue Transmission after beef consumption PrPsc is taken up into Peyer’s patches—(mucousa associated lymphoid Tissue (AKA MALT)—associated with the small intestines T-cells induce its uptake by phagocytic calls that do not destroy the protein Cells leave MALT and enter the lymphatic tissue where they become associated with lymph nodes spleen tonsils Cells can leave the lymphatic system and enter the blood circulatory system via the thoracic duct Also lymphatic tissue is highly enervated so PrPsc can enter nerve cells PrPsc moves up the axon of nerve cells to spinal cord eventually the brain Route of transmission: : MALT to lymphatic system Lymphatic system to blood circulatory system Prevention and Cure Prevention 1. Don’t feed cows to cows 2. Destroy cow population once BSE found 3. Implement sensitive diagnostic tests to identify cows with BSE before they show symptoms There is no good way to destroy PrPsc in living material Cure SO FAR NO CURE!!!! Future treatments might include treatments currently used for Alzheimer’s Disease.