Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
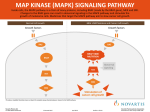
2015 Chabner Colloquium: Collaboration in Cancer Trials is The Society's Official Journal Emerging Therapies to Optimize MAP Kinase Blockade and Intercept Compensatory Signaling Keith T. Flaherty Massachusetts General Hospital Cancer Center, Harvard Medical School Keith T. Flaherty Cancers that harbor BRAF mutations, and RAS mutations to a lesser extent, are considered the most MAP kinase-dependent of tumors. Melanoma is the tumor type that most commonly harbors activating mutations in BRAF (50%) and RAS (25%). Clinical evidence shows that MEK inhibitors produce similar response rates in either subpopulation (∼20%) of patients with BRAF-mutant or RAS-mutant melanoma. BRAF inhibitors, such as vemurafenib and dabrafenib, have a higher therapeutic index in BRAF-mutant melanoma than MEK inhibitors. In a metaanalysis of phase II and III trials in patients with melanoma with BRAF-activating mutations, treatment with a BRAF inhibitor substantially improved overall survival (OS) compared with standard chemotherapy.8 Furthermore, combination therapy with a BRAF inhibitor and a MEK inhibitor resulted in improved OS compared with BRAF inhibition alone.1 In the era of potent immunotherapies for melanoma, oncogene-targeted therapy plays an important therapeutic role for select patients. In patients with BRAF Val600mutated metastatic melanoma, combination therapy with dabrafenib and the MEK inhibitor trametinib significantly improved median OS (25.1 months vs 18.7 months; HR, 0.71; P = 0.0107) and progression-free survival (PFS) (11 months vs 8.8 months; HR, 0.67; P < 0.001) compared with dabrafenib alone.2 Overcoming Treatment Resistance Many patients with BRAF-mutated metastatic melanoma who demonstrate a clinical response to BRAF inhibitor monotherapy maintain a substantial burden of residual disease.3 One explanation for the heterogeneity of response involves acquired resistance to BRAF inhibition. Resistance to BRAF inhibitors readily develops through CRAF. This creates the opportunity to deploy MEK inhibitors with this class of BRAF inhibitors but also highlights the opportunity for RAF inhibitors that inhibit both BRAF and CRAF. The potential for dual BRAF/CRAF inhibitors to have a greater therapeutic index than MEK inhibitors rests, in part, www.STO-online.org on putative MEK-independent functions of CRAF that have yet to be validated in the therapeutic resistance setting. Additional routes of escape from BRAF, MEK, and ERK inhibition in BRAF-mutated melanoma cells involves the activation of a cyclicAMP (cAMP)-dependent signaling network.4 Activation of transcription factors downstream from the MAPK pathway, most notably microphthalmia-associated transcription factor (MITF), also appeared to confer resistance.4 MITF directly regulates the expression of peroxisome proliferator-activated receptor γ coactivator 1-α (PGC1α), the mitochondrial master regulator. Activation of the BRAF/ MAPK pathway in melanoma cells is associated with suppression of MTIF and PGC1α, as well as decreased oxidative metabolism. Conversely, BRAF inhibition renders BRAF-mutated melanocytes addicted to oxidative phosphorylation. This metabolic adaptation appears to limit the efficacy of BRAF inhibitors. Additional mechanisms, such as increased expression of BCL2A1, a lineage-specific antiapoptotic melanoma oncogene, may also facilitate escape from BRAF inhibition in BRAF-mutated melanoma cells. Emerging MAP Kinase Pathway Targets Reactivation of the MAP kinase pathway remains a dominant theme in patients treated with BRAF and MEK inhibitors, suggesting that there is an increasing role for additional agents targeting this pathway. These include both monotherapies as well as oncogenetargeted doublets. Emerging options for MAP kinase pathway antagonism are summarized in Table 1. ERK inhibitors are in late phase I development and appear to produce toxicities similar to MEK inhibitors. However, these agents have yet to be established as being uniquely capable of controlling MAP kinase pathway output and overcoming resistance mechanisms that pertain to MEK inhibitors. HSP90 antagonists are receiving renewed attention as a strategy to © Society for Translational Oncology 2016 2 Emerging Therapies to Optimize MAP Kinase Blockade and Intercept Compensatory Signaling Table 1. Future Directions for MAP Kinase Pathway Antagonism Novel MEK or ERK inhibitors Addition of HSP90 inhibitor Inhibition of RAF dimerization Disrupting scaffolding functions of MAPK components HSP90, heat-shock protein 90; MAPK, MAP kinase. downregulate expression of mutant BRAF and wild-type BRAF and CRAF in combination with kinase inhibitors. Preclinical data support the potential of this approach. New tools are needed to monitor output of the MAP kinase pathway to understand the state of patients’ tumors through the course of therapy and to recognize the points at which new agents or alterations in treatment schedules are needed. Single-cell, bulk tumor, and noninvasive imaging strategies are all in development and may facilitate clinical research in this area. Future Directions Using BRAF-mutant melanoma as the prototype, the goal is to extend newly tested treatment principles to patients with other BRAF-mutant tumor types. For instance, BRAF inhibitor-based treatment regimens such as vemurafenib monotherapy and combination dabrafenib/trametinib have shown activity in patients with BRAF-mutant colorectal cancer.5,6 In the future, the use of biomarkers, drug-sensitivity testing (in vivo or ex vivo), and retesting of serial biopsies after lead-in therapy may guide the selection of immunotherapy or oncogene-targeted therapy at each stage of disease. Financial Disclosures Dr. Flaherty discloses the following financial relationships: Consultant/advisory role: GSK, Novartis, Roche, Sanofi, Merck, Momenta, and Raze. Ownership interests: Clovis and Loxo. References 1. Ugurel S, et al. 2016. In press. 2. Long G V, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet (London, England). 2015;386(9992):444-451. 3. Sosman JA, Kim KB, Schuchter L, et al. Survival in BRAF V600-mutant advanced mela- noma treated with vemurafenib. N Engl J Med. 2012;366(8):707-714. BRAF-mutant metastatic colorectal cancer. J Clin Oncol. 2015;23(Suppl 3); Abstract TPS790. 4. Johannessen CM, Johnson LA, Piccioni F, et al. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature. 2013;504(7478):138-142. 6. Corcoran RB, Falchook GS, Infante JR, et al. BRAF V600 mutant colorectal cancer expansion cohort from the phase I/II clinical trial of BRAF inhibitor dabrafenib (GSK2118436) plus MEK inhibitor trametinib (GSK1120212). J Clin Oncol. 2012;30 (Suppl): Abstract 3. 5. Kopetz S, McDonough S, Morris VK, et al. S1406: Randomized phase II study of irinotecan and cetuximab with or without vemurafenib in © Society for Translational Oncology 2016 OTncologist he ®