Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
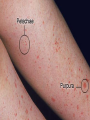
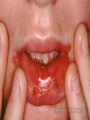
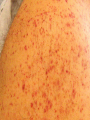
Disseminated Intravascular Coagulation Thrombotic microangiopathy refers to a heterogeneous group of conditions, including disseminated intravascular coagulation (DIC), that result in consumption of clotting factors, platelets, and anticoagulant proteins. Consequences of this process include widespread intravascular deposition of fibrin, leading to tissue ischemia and necrosis, a generalized hemorrhagic state, and hemolytic anemia. Clinical Manifestations DIC accompanies a severe systemic disease process, usually with shock. Bleeding frequently first occurs from sites of venipuncture or surgical incision. The skin may show petechiae and ecchymoses. Tissue necrosis may involve many organs and can be seen as infarction of large areas of skin, subcutaneous tissue, or kidneys. Anemia caused by hemolysis may develop rapidly, owing to microangiopathic hemolytic anemia. Laboratory Findings There is no well-defined sequence of events. Certain coagulation factors (factors II, V, and VIII, and fibrinogen) and platelets may be consumed by the ongoing intravascular clotting process, with resultant prolongation of the prothrombin, partial thromboplastin, and thrombin times. Platelet counts may be profoundly depressed. The blood smear may contain fragmented and burr- and helmet-shaped red blood cells (schistocytes). In addition, because the fibrinolytic mechanism is activated, fibrinogen degradation products (FDPs, D-dimers) appear in the blood. The D-dimer is formed by fibrinolysis of a crosslinked fibrin clot. The D-dimer assay is as sensitive as the FDP test and more specific for activation of coagulation and fibrinolysis Treatment The 1st 2 steps in the treatment of DIC are the most critical: (1) treat the trigger that caused DIC and (2) restore normal homeostasis by correcting the shock, acidosis, and hypoxia that usually complicate DIC. If the underlying problem can be controlled and the patient stabilized, bleeding quickly ceases, and there is improvement of the abnormal laboratory findings. Blood components are used for replacement therapy in patients with hemorrhage and may consist of platelet infusions (for thrombocytopenia), cryoprecipitate (for hypofibrinogenemia), and/or fresh frozen plasma (for replacement of other coagulation factors and natural inhibitors). In DIC associated with sepsis, a controlled trial of drotrecogin alpha (activated protein C concentrate [APC]) in adults with sepsis showed a statistically significant survival advantage in those treated with APC. Clinical trials using protein C concentrate in purpura fulminans and APC in children with sepsis syndrome have not shown a statistically significant improvement. The role of heparin in DIC is limited to patients who have vascular thrombosis in association with DIC or who require prophylaxis because they are at high risk for venous thromboembolism. The prognosis of patients with DIC is primarily dependent on the outcome of the treatment of the primary disease and prevention of end-organ damage Idiopathic (Autoimmune) Thrombocytopenic Purpura The most common cause of acute onset of thrombocytopenia in an otherwise well child is (autoimmune) idiopathic thrombocytopenic purpura (ITP). Pathogenesis Why some children develop the acute presentation of an autoimmune disease is unknown. The exact antigenic target for most such antibodies in most cases of childhood acute ITP remains undetermined. although in chronic ITP most patients demonstrate antibodies against the platelet glycoprotein complexes. After binding of the antibody to the platelet surface, circulating antibody-coated platelets are recognized by the receptor on splenic macrophages, ingested, and destroyed. Most common viruses have been described in association with ITP, including Epstein-Barr virus and HIV. Epstein-Barr virus-related ITP is usually of short duration and follows the course of infectious mononucleosis. Clinical Manifestations The classic presentation of ITP is a previously healthy 1-4 yr old child who has sudden onset of generalized petechiae and purpura. The parents often state that the child was fine yesterday and now is covered with bruises and purple dots. Often there is bleeding from the gums and mucous membranes, particularly with profound thrombocytopenia There is a history of a preceding viral infection 1-4 wk before the onset of thrombocytopenia. Findings on physical examination are normal, other than the finding of petechiae and purpura. Splenomegaly, lymphadenopathy, bone pain, and pallor are rare. Outcome Severe bleeding is rare (<3% of cases in 1 large international study). In 70-80% of children who present with acute ITP, spontaneous resolution occurs within 6 mo. Therapy does not appear to affect the natural history of the illness. Fewer than 1% of patients develop an intracranial hemorrhage. There is no evidence that therapy prevents serious bleeding. Approximately 20% of children who present with acute ITP go on to have chronic ITP. The outcome/prognosis may be related more to age, as ITP in younger children is more likely to resolve whereas the development of chronic ITP in adolescents approaches 50%. Laboratory Findings Severe thrombocytopenia is common, and platelet size is normal or increased, reflective of increased platelet turnover. In acute ITP, the hemoglobin value, white blood cell (WBC) count, and differential count should be normal. Hemoglobin may be decreased if there have been profuse nose bleeds or menorrhagia. Bone marrow examination shows normal granulocytic and erythrocytic series, with characteristically normal or increased numbers of megakaryocytes. Some of the megakaryocytes may appear to be immature and are reflective of increased platelet turnover. Indications for bone marrow aspiration/biopsy include an abnormal WBC count or differential or unexplained anemia as well as findings on history and physical examination suggestive of a bone marrow failure syndrome or malignancy. Other laboratory tests should be performed as indicated by the history and physical examination. In adolescents with new-onset ITP, an antinuclear antibody test should be done to evaluate for SLE. Treatment There are no data showing that treatment affects either short- or long-term clinical outcome of ITP. Many patients with new-onset ITP have mild symptoms, with findings limited to petechiae and purpura on the skin, despite severe thrombocytopenia. Compared with untreated control subjects, treatment appears to be capable of inducing a more rapid rise in platelet count to the theoretically safe level, although there are no data indicating that early therapy prevents intracranial hemorrhage. Antiplatelet antibodies bind to transfused platelets as well as they do to autologous platelets. Thus, platelet transfusion in ITP is usually contraindicated unless life-threatening bleeding is present. Initial approaches to the management of ITP include the following: 1 No therapy other than education and counseling of the family and patient for patients with minimal, mild, and moderate symptoms, as defined earlier. 2 Intravenous immunoglobulin (IVIG). IVIG at a dose of 0.8-1.0 g/kg/day for 1-2 days induces a rapid rise in platelet count (usually >20 x 109/L) in 95% of patients within 48 hr. 3.Prednisone. Corticosteroid therapy has been used for many years to treat acute and chronic ITP in adults and children. Doses of prednisone of 1-4 mg/kg/24 hr appear to induce a more rapid rise in platelet count than in untreated patients with ITP. 4.Intravenous anti-D therapy. For Rh positive patients, IV anti-D at a dose of 50-75 ?g/kg causes a rise in platelet count to >20 x 109/L in 80-90% of patients within 48-72 hr. In the special case of intracranial hemorrhage, multiple modalities should be used, including platelet transfusion, IVIG, high-dose corticosteroids, and prompt consultation by neurosurgery and surgery. The role of splenectomy in ITP should be reserved for 1 of 2 circumstances. The older child (≥4 yr) with severe ITP that has lasted >1 yr (chronic ITP) and whose symptoms are not easily controlled with therapy is a candidate for splenectomy. Splenectomy must also be considered when life-threatening hemorrhage (intracranial hemorrhage) complicates acute ITP, if the platelet count cannot be corrected rapidly with transfusion of platelets and administration of IVIG and corticosteroids. Chronic Idiopathic Thrombocytopenic Purpura Approximately 20% of patients who present with acute ITP have persistent thrombocytopenia for >12 mo and are said to have chronic ITP. At that time, a careful re-evaluation for associated disorders should be performed, especially for autoimmune disease, such as SLE; chronic infectious disorders, such as HIV; and non immune causes of chronic thrombocytopenia, such as type 2B and platelet-type von Willebrand disease, X-linked thrombocytopenia, autoimmune lymphoproliferative syndrome, common variable immunodeficiency syndrome.