Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Cardiac contractility modulation wikipedia , lookup
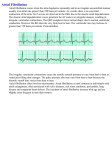
Electrocardiography wikipedia , lookup
Quantium Medical Cardiac Output wikipedia , lookup
Jatene procedure wikipedia , lookup
Arrhythmogenic right ventricular dysplasia wikipedia , lookup
Ventricular fibrillation wikipedia , lookup
Physiol Rev 94: 609 – 653, 2014 doi:10.1152/physrev.00022.2013 CARDIAC POTASSIUM CHANNEL SUBTYPES: NEW ROLES IN REPOLARIZATION AND ARRHYTHMIA Nicole Schmitt, Morten Grunnet, and Søren-Peter Olesen The Danish National Research Foundation Centre for Cardiac Arrhythmia, Department of Biomedical Sciences, University of Copenhagen, Copenhagen, Denmark; Acesion Pharma, Copenhagen, Denmark; and Lundbeck A/S, Valby, Denmark L I. II. III. IV. V. INTRODUCTION ION CHANNELS IN ATRIAL AND... MECHANISMS AND ETIOLOGY... PHARMACOLOGICAL TOOLS... CONCLUSIONS AND PERSPECTIVES 609 611 625 635 639 I. INTRODUCTION The heart functions as a mechanical pump ensuring a continuous systemic and pulmonary blood supply. The circulation provides the essential transport of nutrients, antibodies, hormones, and other chemical mediators, as well as the exchange of gases and removal of waste products. Under normal physiological, i.e., nondiseased conditions, the heart works steadily and uninterruptedly, only changing rate according to changes in systemic needs. A human heart thus performs ⬃100,000 successful and coordinated contractions every day. These contractions are controlled by electrical signals, known as action potentials, which trigger an intracellular signaling pathway referred to as the excitation-contraction coupling leading to the muscle contraction (41). The tight electrical control of cardiac contractions also means that imbalances can lead to disturbances in the heart rhythm. In fact, under certain conditions, a single inappropriate electrical signal can obliterate proper cardiac function and ultimately develop into sudden cardiac arrest. Any deviation from the normal stable heart rhythm is categorized as an arrhythmia (from Greek a ⫹ rhythmos ⫽ loss of rhythm). Arrhythmias are often diagnosed based on irreg- ularities of the surface electrocardiogram (ECG), which macroscopically depicts the electrical activity of the heart. The ECG is divided into different phases (FIGURE 1) that reflect the depolarization and repolarization of atria and ventricles. Arrhythmias span the entire spectrum from the simplest isolated event of a diminutive palpitation to uncoordinated chaotic signaling recognized as fibrillations. The latter is especially dangerous in a context where fibrillation propagates in the ventricles. Unless quickly terminated, ventricular fibrillation can result in sudden cardiac death, as it invalidates coordinated pumping activity. Arrhythmias are, however, unusual events in young and middle-aged people, which is comforting and also indicates the presence of a broad safety margin and tight control of the complicated and multifaceted processes of electrical signaling and excitation-contraction coupling. In combination with partly overlapping functions for a number of the proteins involved in these mechanisms, this ensures a high level of fidelity and stability in the cardiac system. The high lipophilicity of cell membranes, including those of cardiac myocytes, provides an effective separation barrier between the cell interior and the extracellular surroundings. The cell membrane is impermeable to the hydrophilic ions, but special transmembrane proteins such as ion channels and transporters enable the different ion species to permeate selectively, and they form the basis for a controlled 0031-9333/14 Copyright © 2014 the American Physiological Society 609 Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 Schmitt N, Grunnet M, Olesen S-P. Cardiac Potassium Channel Subtypes: New Roles in Repolarization and Arrhythmia. Physiol Rev 94: 609 – 653, 2014; doi:10.1152/physrev.00022.2013.—About 10 distinct potassium channels in the heart are involved in shaping the action potential. Some of the K⫹ channels are primarily responsible for early repolarization, whereas others drive late repolarization and still others are open throughout the cardiac cycle. Three main K⫹ channels drive the late repolarization of the ventricle with some redundancy, and in atria this repolarization reserve is supplemented by the fairly atrial-specific KV1.5, Kir3, KCa, and K2P channels. The role of the latter two subtypes in atria is currently being clarified, and several findings indicate that they could constitute targets for new pharmacological treatment of atrial fibrillation. The interplay between the different K⫹ channel subtypes in both atria and ventricle is dynamic, and a significant up- and downregulation occurs in disease states such as atrial fibrillation or heart failure. The underlying posttranscriptional and posttranslational remodeling of the individual K⫹ channels changes their activity and significance relative to each other, and they must be viewed together to understand their role in keeping a stable heart rhythm, also under menacing conditions like attacks of reentry arrhythmia. SCHMITT, GRUNNET, AND OLESEN lated in shape, with a less pronounced plateau phase and a more sliding repolarization phase. Action potentials recorded from sinoatrial and atrioventricular nodes exhibit a third kind of shape with a slow onset enabling pacemaker activity. This is accomplished by a sliding resting membrane potential that will elicit spontaneous activity when depolarization is sufficient to reach the threshold for initiation of action potentials. Keeping in mind that ion channels confer the unique signature of action potentials, it follows that a thorough understanding of the regulation and interaction between these channels is a prerequisite for the perception of cardiac function in both a physiological and a pathophysiological context. Representative examples of action potentials obtained from human atria and ventricles are shown in FIGURE 2. QRS Complex R PQ Interval T Q S QT Interval FIGURE 1. Electrocardiogram (ECG) hallmarks. ECG from an individual in normal sinus rhythm. The schematic drawing below (adapted from Wikimedia Commons) illustrates the phases of the ECG. The P-wave represents depolarization of the atria, the QRS complex the depolarization of the ventricles, and the T-wave the repolarization of the ventricles. electrical communication across the cell membrane as the ions carry electrical charges. Fast transient changes in ion permeability generate action potentials and thereby excitability. This is primarily achieved by delicate control of the opening and closing of a number of different ion channels in various parts of the heart. Cardiac action potentials are different from their neuronal counterparts by having an extended plateau phase that will last for several hundred milliseconds in humans and thereby secure the necessary Ca2⫹ dynamic for appropriate contractions and time for the heart chambers to empty their blood volume. The morphology of the cardiac action potential varies in shape and duration according to the location in the heart (reviewed in Ref. 305). Ventricular action potentials have a relative stable plateau phase and what is recognized as a domelike shape. The exact action potential shape varies to a minor but significant degree in different parts of the ventricle and throughout the ventricular wall. Furthermore, the shape varies over time depending on heart rate, as a fast heart rate speeds up repolarization. Atrial action potentials appear more triangu- 610 Ion channels are, as stated, of paramount significance for electrical signaling in excitable cells. A thorough insight into the function and regulation of cardiac ion channels is therefore important for the understanding of cardiac physiology and pathophysiology. This review has consequently been devoted to giving a detailed description of cardiac ion channel function and regulation with special emphasis on repolarizing K⫹ channels and their role in Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 P ST Segment PQ Segment The stability of cardiac electrical signaling is secured by some degree of redundancy in combination with the tight regulation of ion channels as stated above. The various K⫹ channels involved in repolarization of the action potential represent a good example of such partial redundancy. The most thoroughly investigated example is from the ventricles, where three different K⫹ currents participate in the same phase of repolarization. These currents are called IKr, IKs, and IK1, and albeit different in nature, due to different biophysical properties these currents all have a profound and overlapping impact on repolarization of the action potential. This phenomenon has been described as the “repolarization reserve” (364) to emphasize the partly overlapping function and, in this context, the redundancy of the system (FIGURE 2). Various K⫹ channels also participate in the repolarization of atrial action potentials, so there is an equivalent atrial repolarization reserve, including a number of additional K⫹ channel subtypes (FIGURE 2). Some K⫹ channels are believed to be exclusively present in the atria and therefore play important roles for the triangulated shape of atrial action potentials compared with their more domelike ventricular counterparts. Examples of K⫹ channels selectively expressed in the atria encompass Kir3.1/ Kir3.4 as responsible for IKACh, KV1.5 as responsible for IKur, and, more recently, Ca2⫹-activated K⫹ channels accountable for IKCa and two-pore domain K⫹ channels K2P underlying Ileak (51, 102, 122, 132, 166, 250, 302, 471). Due to their limited distribution, these channels have gained significant interest for many years in the continuing effort to find a treatment for atrial fibrillation without concomitant ventricular adverse effects. CARDIAC POTASSIUM CHANNELS AND ARRHYTHMIA Sinus node Right atrium Left atrium Atrioventricular node Left ventricle Right ventricle –50 0 mV mV –50 01 2 100 ms 3 4 mV 01 100 ms 2 Ito 3 4 IKs IKr IK1 IKATP IKur IKACh IKCa ITASK arrhythmia. To obtain better perspectives on the nature of arrhythmias and the role of ion channels in this context, we include an introduction to the mechanisms and etiology of arrhythmias. Finally, we cover pharmacological treatments of arrhythmias and the general application of pharmacological tools to obtain better physiological understanding of the heart. II. ION CHANNELS IN ATRIAL AND VENTRICULAR CARDIOMYOCYTES The different cardiac ion channels play well-defined roles in shaping the action potential. Although the overall principle is the same in all mammalian cells, the role of repolarizing currents differs significantly. In the heart, it is widely dic- tated by the great differences in heart rate among species. Humans have a resting heart rate of ⬃60 beats/min (bpm), whereas mice have a heart rate of ⬃600 bpm, which requires repolarizing K⫹ channels with different kinetics. Within the same species, the ion channel expression and thereby the shape and length of the action potential also varies significantly in subregions of the heart, as shown in FIGURE 2. These local differences, especially between atria and ventricles, but also in sinoatrial (SA) and atrioventricular (AV) node, the conduction system and across the ventricular wall, are crucial for the function of these subregions and for the stability of the heart rhythm. The following section gives a short account of the ion channels responsible for shaping action potential in atria and Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org 611 Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 0 mV FIGURE 2. Cardiac action potential and repolarizing currents that underlie the repolarization reserve in atria and ventricles. Original traces from an action potential recording in human atrial trabecula (left) and ventricular septum (right). Note that the atrial resting membrane potential (RMP) is more positive than the ventricular RMP (indicated by the red dotted line). K⫹ current contributions to the different phases (0 – 4) of the action potential are shown below with an approximate physiological time course. The current amplitudes are arbitrary and do not reflect their relative size. SCHMITT, GRUNNET, AND OLESEN ventricle with focus on potassium channels. A more detailed description is included for two subtypes of K⫹ channels that traditionally have received relatively little attention. These are the Ca2⫹-activated K⫹ channels and two-pore domain K⫹ channels. We further elaborate on the role of accessory subunits, posttranslational modifications, and epigenetic regulation for the major repolarizing potassium currents. A. Cardiac Action Potential: The Classic Orchestra 1. Phase 0 The abrupt shift in membrane potential in a positive direction (depolarization) that initiates the systolic period is called phase 0 and is driven by influx of Na⫹, INa, through the NaV1.5 channel encoded by the gene SCN5A. The channels are rather insensitive to tetrodotoxin (TTX), which blocks many neuronal NaV channels (126, 482). The NaV1.5 current is by far the dominating Na⫹ current in the human heart, but TTX-sensitive currents have been reported (99, 365, 393, 502). Discrete expression patterns have been shown by immunohistochemistry for NaV1.1, NaV1.5 channels open at potentials positive to ⫺55 mV, show a steep voltage dependency, and activate very quickly (126). In addition to the fast peak current, NaV1.5 channels also conduct a persistent current due to slow or incomplete inactivation at intermediate potentials (23). Although this persistent or window current only makes up 0.1–1% of the total current (441), it can significantly impact the action potential morphology and arrhythmogenesis (37, 451). The NaV1.5 channels are encoded by the same gene throughout the heart, but functional differences exist between atria and ventricle. The voltage for half-inactivation of NaV1.5 channels is more negative in atria compared with ventricles in canine and guinea pig (59, 243), and the resting membrane potentials differ between the two regions, which impacts Na⫹ channel availability and conduction velocity. 2. Phase 1 The initial depolarization is followed by a transient repolarization termed phase 1, which is most prominent in atria and the ventricular epicardium. This early repolarization is caused by inactivation of NaV1.5 channels in combination Table 1. Cardiac potassium currents Current Pore Forming ␣-Subunit Gene Other Channel Names Topology Phase Ito, fast KV4.3 KCND3 6 TM tetramer 1 Ito, slow IKur IKr KV1.4 KV1.5 KV11.1 KCNA4 KCNA5 KCNH2 6 TM tetramer 6 TM tetramer 6 TM tetramer 1 1 (1–3) 3 (3–4) IKs Kv7.1 KCNQ1 6 TM tetramer 3 (2–4) IK1 IKAch IKATP Kir2.1 Kir3.1/Kir3.4 Kir6.1 KCNJ2 KCNJ3/KCNJ5 KCNJ11 2 TM tetramer 2 TM tetramer 6 TM tetramer 3 (3–4) 3 0–3 Ileak IKCa K2P3.1 KCa2.x KCNK3 KCNN1-KCNN3 4 TM dimer 6 TM tetramer 0–4 0–4 ERG1, hERG, LQT2 KvLQT1, KCNQ1, LQT1, SQT2 GIRK2 GIRK1/GIRK4 TASK-1 SK1, SK2, SK3 Comment Reference Nos. Additional obligatory subunit KChIP2 Reviewed in 311 Atria-specific Additional obligatory subunit KCNE1 Atria-specific Additional obligatory subunit SUR2A TM, transmembrane segment. 612 Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org 133, 415 Reviewed in 359 Reviewed in 433 Reviewed in 266, 464 98, 285 Reviewed in 113 Reviewed in 24 Reviewed in 166 Reviewed in 153 Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 The cardiac action potential is divided into five distinct phases (phase 0 – 4, as indicated below the action potentials in FIGURE 2). The underlying endogenous currents and their molecular correlates are described below. TABLE 1 gives an overview of potassium currents, their pore-forming ion channel subunits, the principle channel topology, and the genes encoding the channels. For stringency, we follow the nomenclature recommendations of the International Union of Basic and Clinical Pharmacology (IUPHAR) (145, 450, 483), although trivial channel names, included in TABLE 1, are widely used in the field. NaV1.2, NaV1.3, NaV1.4, and NaV1.6 (encoded by the genes SCN1A, SCN2A, SCN3A, SCN4A, and SCN6A, respectively) (210, 452). Furthermore, the NaV1.8 channel protein encoded by SCN10A was reported to be expressed in working myocardium and the cardiac conduction system (475). Others, however, have found its expression restricted to intracardiac neurons (436), and the impact of NaV1.8 on the cardiac sodium current is debated (256). Whereas NaV1.5 is the main depolarizing current in atria, conduction system, and ventricle, it is absent in the core of the nodes where CaV currents are suggested to serve this function, albeit with slower kinetics (271, 305, 323). CARDIAC POTASSIUM CHANNELS AND ARRHYTHMIA Selectively in atria, an additional ultra-rapid current called IKur contributes to early repolarization. IKur is conducted by KV1.5 potassium channels, and silencing KV1.5 in human atrial myocytes by antisense oligonucleotides causes a major reduction of the IKur current (122, 359, 397). Due to its slow and partial inactivation, IKur plays a role during all phases 1–3, being partly responsible for the triangular action potential shape. 3. Phase 2 The long-lasting depolarized plateau in phase 2 of the cardiac action potential is a prerequisite for excitation-contraction coupling and is caused by a balance between outward K⫹ currents and inward currents through voltagegated Ca2⫹ channels. The latter are located in the t tubules, and the Ca2⫹ influx through these channels activates ryanodine receptors located just below in the sarcoplasmic reticulum. The ensuing Ca2⫹-dependent Ca2⫹ release from intracellular stores triggers muscle contraction (41). There are two types of CaV channels displaying different expression patterns: the CaV1.2 (L-type, gene CACNA1C) ubiquitously expressed in the heart, and the CaV3 (T-type) suggested to contribute to the pacemaker function in the nodes (30, 309, 323, 398, 496). CaV3 exists in three isoforms: CaV3.1, 3.2, and 3.3. CaV3 channels activate at potentials positive to ⫺50 mV and inactivate quickly, whereas CaV1.2 channels require potentials positive to ⫺30 mV and inactivate slowly, making them the key driver of phase 2 (30, 339). Phase 2 is terminated by a voltage- and Ca2⫹/calmodulin-dependent slow inactivation of CaV1.2 combined with the simultaneous activation of K⫹ channels (40, 97). To a lesser extent, the Ca2⫹ influx in the early part of phase 2 is also transported by the electrogenic Na⫹/Ca2⫹ exchanger encoded by the SLC8A1 gene. The stoichiometry of this cotransporter is 3 Na⫹ for 1 Ca2⫹, which results in a net movement of a positive charge with a reversal potential around ⫺30 mV causing influx of Ca2⫹ and extrusion of Na⫹ during phase 2 in normal hearts (20, 41, 273). 4. Phase 3 Phase 3 represents the repolarization from plateau phase to resting membrane potential. The repolarization is caused by the balance between inactivating Ca2⫹ current and rising K⫹ channels tilting quickly towards the latter. In humans and other large mammals, three different potassium currents called IKr, IKs, and IK1 are mainly responsible for repolarization. They have diverse but partly overlapping functions, and to stress the latter they have often been called the “repolarization reserve” (FIGURE 2) (364). IKr is conducted by the channel protein KV11.1, also known as ERG1 or just hERG for the human variant. The KV11.1 channel is the gene product of KCNH2, and it exists in several splice variants called KV11.1a, KV11.1b, and KV11.1-uso (233). Functional IKr is likely to consist of a mixture of both KV11.1a and KV11.1b (232, 255). The presence of accessory -subunits might further be necessary to recapitulate native IKr, and two of these called KCNE1 and KCNE2 have been described to interact with KV11.1 (2, 280). The proposed interaction with KCNE1 has been substantiated by immunoprecipitations from horse heart, indicating a direct interaction (124), whereas the interaction with KCNE2 remains controversial (116). IKs is conducted by the pore forming ␣-subunit KV7.1 (KCNQ1) in a complex with the -subunit KCNE1 (also known as mink or IsK) (29, 266, 376). KV7.1 channels can potentially interact with all -subunits in the KCNE family (KCNE1–5) (279), since although at different levels, all five subunits are expressed in the heart and modulate the function of KV7.1 in vitro. IK1 is conducted via Kir2.x proteins. This channel family consists of four members Kir2.1 (IRK1/KCNJ2), Kir2.2 (IRK2/KCNJ12), Kir2.3 (IRK3/KCNJ4), and Kir2.4 (IRK4/ KCNJ14) (47, 53, 226, 413, 421). Intriguingly, the partly overlapping function of these three currents driving phase 3 repolarization occurs on the basis of very different gating properties. The voltage-gated current IKr is characterized by a relatively fast activation upon depolarization, but as the inactivation is another 10-fold faster, it renders the channel virtually nonconductive during phases 0 –2 of the action potential (346). The opposite is true in phase 3, where the initial repolarization will release IKr from inactivation and reopen the channel. The subsequent deactivation or closure of the channel is a slow process allowing IKr to conduct current during both phase 3 and the early part of the diastolic phase 4 of the action potential. Although IKs is also voltage-gated, it displays very different biophysical properties. IKs opens at potentials positive to ⫺20 mV and activates slowly. Both the slow activa- Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org 613 Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 with activation of transient voltage-gated K⫹ currents (Ito) that can further be divided into Ito,fast and Ito,slow (470). Both are activated at potentials above ⫺30 mV and show inactivation. The differences in the two Ito components exhibit in their kinetics of inactivation and recovery from inactivation (Ito,fast: inactivation ⫽ recovery ⫽ 20 –100 ms; Ito,slow: inactivation ⫽ hundreds of ms, recovery ⫽ seconds) (311, 335). The pore-forming subunit of Ito,fast is KV4.2 and KV4.3 channels, encoded by the genes KCND2 and KCND3, respectively. Additionally, the accessory subunits KChIP2 and DPP6 are necessary to recapitulate native Ito,fast (165). In large mammals, Ito,fast is primarily formed by KV4.3 ⫹ KChIP2, and the latter shows a stronger epicardial than endocardial expression, causing the Ito gradient across the ventricular wall (367, 399). The K⫹ channel constituting Ito,slow is KV1.4 (KCNA4) (311). Functionally, the fast repolarization in phase 1 contributes to increasing the driving gradient for Ca2⫹ into the cardiac myocytes. SCHMITT, GRUNNET, AND OLESEN In addition to the “repolarization reserve currents” IKr, IKs, and IK1, other potassium currents can have a significant impact on repolarization of cardiac action potentials. Especially important for the atria are IKur (see phase 1) and IKACh. IKACh is generally believed to be atria specific, and endogenous IKAch channels are composed of Kir3.1/Kir3.4 (or GIRK1/GIRK4) proteins encoded by KCNJ3 and KCNJ5 genes. IKAch is directly activated by interaction with the ␥ part of Gi proteins connected to muscarinic receptors (178). Release of acetylcholine following vagal stimulation leads to activation of the cardiac muscarinic receptors. Kir3.1/ Kir3.4 channels play an important role in the sinoatrial node, where an increased K⫹ conductance will give rise to a slower depolarization of the pacemaker potential and a reduction of the heart rate (281, 358). Furthermore, Kir3.1/Kir3.4 channels are important for atrial repolarization. Activation of the channels causes a shortening of the atrial action potential. The channel is also activated by adenosine and seems via this mechanism to be able to also repolarize both atrial (445) and ventricular cardiomyocytes (246). Finally, IKATP should be mentioned as a potentially important cardiac repolarizing current. The channel is composed of the pore-forming ␣-subunit Kir6.2 (KCNJ11) in association with the -subunit SUR2A (ABCC9) (59a, 124a). The channel is abundant throughout the heart, but generally closed due to ATP inhibition at the intracellular surface. Under normal physiological conditions, the intracellular 614 ATP level is sufficiently high to inhibit the channels, but during hypoxia and ischemia, the intracellular ATP level drops and may result in an activation of Kir6.2 channels (189a). Kir6.2 channels have a membrane topology similar to Kir2.x channels conducting IK1, albeit with less rectification, so they can conduct current throughout phases 0 –3 resulting in triangulation of the action potential. 5. Phase 4 Phase 4 describes the interval between full repolarization and the initiation of the next action potential. In this diastolic interval, the cardiac myocytes are at resting membrane potential, and the charge movements are primarily conducted via various K⫹ channels, including Kir2.x and two-pore motif leak channels. Additional K⫹ channels with slow deactivation kinetics, including KV11.1 and KV7.1, also remain open in the initial part of phase 4. These K⫹ currents will counteract possible depolarizing events from delayed afterdepolarizations. However, the inhibitory force of the K⫹ conductances is not enough to withstand massive depolarizing stimuli arising from a new wave of action potentials. Prolongation of the effective refractory period through K⫹ channel inhibition and Na⫹ channel inactivation remains the more efficient mechanism. It is worth mentioning that the duration and degree of hyperpolarization in phase 4 will impact the morphology of the subsequent action potential. The reason is that both Ca2⫹ and Na⫹ channels undergo voltage- and time-dependent release from inactivation. The number of Na⫹ and Ca2⫹ channels that can be engaged in a subsequent action potential will therefore increase with duration and extent of hyperpolarization in phase 4. B. Cardiac Action Potential: New Players In addition to the well-described currents, in recent years Ca2⫹-activated small conductance K⫹ channels (KCa2.x) and various two-pore domain channels (K2P) have proven to be important cardiac K⫹ channels contributing in particular to atrial repolarization. 1. Ca2⫹-activated K⫹ channels For many years, Ca2⫹-activated K⫹ channels were regarded as absent from cardiac tissue or at least not present in cardiomyocytes. There is increasing evidence of an important functional role for some subtypes of Ca2⫹-activated K⫹ channels in the heart. After a brief introduction to the family of Ca2⫹-activated K⫹ channels and the pharmacological tools applied to reveal their function, we will provide an overview of the literature evidence for the significance of these channels in cardiac tissue. Ca2⫹-activated K⫹ channels are divided into three subfamilies originally classified according to single-channel Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 tion kinetics and the right-shifted activation curve are largely caused by the presence of the KCNE1 -subunit. Importantly and in contrast to IKr, IKs barely inactivates (195, 403). These properties enable IKs to build up slowly during phase 2 and to become one of the key K⫹ conductances in phase 3. The third current, IK1, is not endowed with an inherent voltage sensor in the protein (175). However, Kir2.x channels are indirectly voltage sensitive, as the outflow of K⫹ through the channel is accompanied by Mg2⫹ and polyamines such as spermine that will block the channel deep in the pore (120, 164, 258, 432). The more positive the membrane potential becomes, the stronger the inhibition and Kir2.x channels do not conduct K⫹ at potentials more positive than ⫺20 mV, i.e., in phases 0 –2. When phase 3 repolarization has reached a value of approximately ⫺40 mV, the Kir2.x channels will be released from the Mg2⫹/spermine inhibition, and IK1 will contribute particularly to repolarization in the late part of phase 3 (258). IK1 stays open during diastole and is important for setting the resting membrane potential. This is emphasized by the fact that ventricles with high IK1 expression have a resting membrane potential of approximately ⫺80 mV, while the maximum diastolic potential in nodes lacking IK1 is about ⫺50 mV (380). The relative conductances of the three repolarizing currents IKr, IKs, and IK1 during the atrial and ventricular action potential and their partly overlapping functions are illustrated in FIGURE 2. CARDIAC POTASSIUM CHANNELS AND ARRHYTHMIA KCa2.x channels are composed of four ␣-subunits. Each subunit has six transmembrane segments with amino- and carboxy-termini located intracellularly (435) (FIGURE 3B). Cardiac KCa2.x channels can consist of KCa2.1, KCa2.2, and KCa2.3 homotetramers or heterotetramers (425). Functional KCa2.x channels in cardiac tissue have been described in humans, mice, rats, guinea pigs, rabbits, and dogs (101, 102, 245, 295, 328, 426, 471). KCa2.x (and KCa3.x) channels represent a special family of K⫹ channels, since they are voltage insensitive and gated solely by free intracellular conductance resulting in big- (KCa1.1/BK/KCNMA1), intermediate- (KCa3.1/IK/KCNN4), and small-conductance (KCa2.1/SK1/KCNN1, KCa2.2/SK2/KCNN2, KCa2.3/ SK3/KCNN3) potassium channels (39, 161, 449) (FIGURE 3A). To date, only KCa2.x channels have been established as functional in surface membranes of cardiomyocytes, while BK channels have been demonstrated to be important in mitochondrial membranes (17, 37, 38, 444). Since only KCa2.x channels have been associated with arrhythmia, the following description will focus on this channel type. A KCa2.3 (NP_002240) KCa2.2 (NP_067627) Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 KCa2.1 (NP_002239) KCa3.1 (NP_002241) KCa1.1 (NP_001014797) 200 150 100 B 50 0 C nA mV 1 –100 –50 50 100 –15 –15 CaM 2 –15 D % Residual current 100 75 50 IC50 = 91 nM 25 0 10 100 [NS8593] (nM) 1,000 FIGURE 3. Overview of KCa channels. A: phylogenetic tree of KCa channel proteins (GenBank accession numbers to the right). The x-axis denotes amino acid substitution per 100 residues (ClustalW alignment performed with MegAlign, DNASTAR program package). B: topological model of a KCa2.x subunit with calmodulin (CaM) (top) and a tetramer (bottom). C: representative whole cell current recordings of HEK293 cells stably expressing KCa2.3 channels. Both traces represent recordings upon application of 200-ms voltage ramps (every 5 s) from a holding potential of 0 mV to potentials from ⫺100 to ⫹100 mV with 1 M Ca2⫹ in the pipette. Traces were recorded just after break-in (trace 1) and after 60 s (trace 2). [From Grunnet et al. (157). Copyright 2001, with permission from Elsevier.] D: inhibition of KCa2.3 channel activity by the tool compound NS8593. [From Strøbaek et al. (408).] Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org 615 SCHMITT, GRUNNET, AND OLESEN Ca2⫹ concentration ([Ca2⫹]i) (FIGURE 3C). Consequently, KCa2.x channels can integrate changes in secondary messenger activity, i.e., [Ca2⫹]i with changes in membrane K⫹ conductance (449). KCa2.x channels do not form complexes with traditional K⫹ channel -subunits, but constitutively bind calmodulin at the carboxy-terminus of the ␣-subunit. Half-maximal activation appears at ⬃300 nM [Ca2⫹]i, and Ca2⫹ binding is a cooperative event illustrated by the steep Hill coefficient of 4 –5 (468). The Ca2⫹ source for activation of KCa2.x channels is likely L-type CaV channels that are physically coupled to Ca2⫹-activated K⫹ channels in both cardiac and neuronal tissue eventhough Ca2⫹ can also arise from intracellular stores (159, 260). The first indirect evidence for the presence of cardiac KCa2.x channels appeared more than 40 years ago. In a technical report from Edgewood Arsenal, Maryland, apamin was tested in isolated, perfused hearts from rhesus monkeys and mongrel dogs as well as and in intact animals (437). The studies demonstrated clear antiarrhythmic effect without concomitant change in blood pressure (437). These studies were conducted using a bee venom batch in which the different components were not entirely separated, and before apamin was known as a specific ion channel inhibitor (136). It is probably for these reasons that the interesting findings did not lead to follow-up studies for decades. Sporadic indications of a Ca2⫹-activated K⫹ current were occasionally reported (115), but it was not until 1999 that 616 It has further been demonstrated that pharmacological inhibition of IKCa is antiarrhythmic in species such as rat, guinea pig, and rabbit in a model applying isolated perfused hearts. Additional antiarrhythmic effect of KCa2.x inhibition was demonstrated in acute burst-induced AF in vivo in rats (102). KCa2.x channel inhibition can also prolong the atrial effective refractory period in rats in vivo. This prolongation is associated with an antiarrhythmic effect (396). These effects were additionally confirmed in a more clinically relevant setting using aged spontaneously hypertensive rats (101). These rats exhibited atrial remodeling and thereby increased susceptibility to atrial tachyarrhythmias (75). The efficacy of KCa2.x channel inhibition and the observed antiarrhythmic effect were similar in hypertensiveinduced atrial remodeled rats and in young normotensive Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 Specific pharmacological tools have successfully been used to address KCa2.x channel function, with the bee venom apamin as the gold standard. Apamin inhibits all three subtypes of KCa2.x channels at nanomolar concentrations, although with different potency (156, 409). The toxin does not show affinity for the other members of the Ca2⫹-activated K⫹ channel family or for Ca2⫹, Na⫹, and Cl⫺ channels. Therefore, apamin has been considered a fingerprint or state-of-the-art inhibitor of KCa2.x channels. Whereas in noncardiac tissue such as neurons, skeletal muscle, hepatocytes, and T lymphocytes, apamin is used broadly as a tool to address the physiological roles of KCa2.x channels, the experiences from using the large apamin molecule as a tool compound on cardiac tissue are less convincing (28, 46, 101, 191, 354, 396). A number of alternatives to apamin exist, including peptide toxins such as scorpion toxins scyllatoxin and tamapin (406, 465), and small organic molecules such as tubocurarine, UCL1684, or N-(pyridin-2-yl)4-(pyridin-2-yl)thiazol-2-amine (138, 248, 366, 465). A completely different class of selective KCa2.x channel inhibitors was introduced by the small organic compound NS8593, which has a different binding site than apamin (194, 400, 408) (FIGURE 3D). NS8593 acts as a negative gating modifier that decreases the Ca2⫹ sensitivity by rightward shifting the Ca2⫹-activation curve (408). the first convincing data for cardiac KCa2.x channels were presented. Wang et al. (444) reported evidence for KCa2.3 subunits and corresponding IKCa currents in a myocyte cell line derived from rat ventricles (444). In 2003, Xu et al. (471) demonstrated apamin-sensitive currents recorded from human and mouse atrial myocytes, while almost no IKCa could be measured in ventricular myocytes from mice. The results were obtained with a remarkably low apamin concentration of only 50 pM (471). In 2005, Tuteja et al. (426) demonstrated biochemical evidence in mouse hearts for all three KCa2.x channel subtypes. Here KCa2.1 and KCa2.2 subunits revealed an atria-selective distribution (426). In 2007, the first circumstantial evidence of a relationship between KCa2.x channel and arrhythmia was provided (328). This study demonstrated increased membrane trafficking of KCa2.2 channels to pulmonary vein sleeves after burst pacing and a corresponding progressive shortening of action potentials as a consequence of increased IKCa. In 2009, more direct evidence of IKCa association to AF was given (245). Prolongation of atrial action potentials was observed in KCa2.2 knockout mice, and this was associated with a counterintuitive proarrhythmic effect. A likely explanation for the latter could be the sustained and sliding repolarization observed in the knockout mice (triangulation), which is associated with increased propensity for inducible early afterdepolarizations (EAD) under certain conditions. As mentioned, these findings were surprising since prolongation of the atrial effective refractory period is normally considered antiarrhythmic (304). Later in 2009, a new study failed to show any functional role for cardiac KCa2.x channels in rat, dog, and humans under normal physiological conditions, since 100 nM apamin did not affect atrial or ventricular action potential duration in single cardiomyocytes or multicellular preparations, even in conditions with high [Ca2⫹]i (295). One year later, the first clinical link between KCa2.x channels and atrial fibrillation (AF) appeared. A genome-wide association study including 1,355 individuals with lone AF and 12,844 unaffected controls concluded that common gene variants in the area comprising KCNN3 (the gene encoding KCa2.3) are connected to AF (118). CARDIAC POTASSIUM CHANNELS AND ARRHYTHMIA controls (101) (FIGURE 3C). Information regarding functional KCa2.x channels in remodeled human hearts is sparse. Two articles written in Chinese indicate the presence of apamin-sensitive currents in cardiomyocytes isolated from AF patients in sinus rhythm at the time of the recordings. Furthermore, the apamin-sensitive currents are suggested to be upregulated in persistent AF patients (244, 484). A new study comparing human atrial tissue from patients in sinus rhythm and in AF shows that the KCa2.x channel inhibitor NS8593 prolongs the atrial action potential and refractory period (L. Skibsbye, C. Poulet, J. G. Diness, B. H. Bentzen, L. Yuan, U. Kappert, K. Matschke, E. Wettwer, U. Ravens, M. Grunnet, T. Christ, and T. Jespersen, unpublished data). Two-pore domain K⫹ (K2P) channel proteins are a relatively new addition to the superfamily of potassium channels (333). These channels are widely expressed and are major determinants of background K⫹ conductance in the cells in which they are present. They set the negative resting potential of eukaryotic cells under a range of normal and pathophysiological conditions (166, 315). This section will provide a brief introduction to the family of K2P channels, followed by an account of the possible significance of these channels in human atria. Fifteen K2P channel subtypes are known to date, divided into subclasses by homology (FIGURE 4A). Functionally, they lack voltage dependence, but are instead regulated by lipids, temperature, pH, oxygen, membrane stretch, anesthetics, and second messengers (166, 237). Their widely used trivial names arise from functional attributes such as tandem of “P domains in a weak inward-rectifier K⫹ channel” (TWIK), “TWIK-related channel” (TREK), or “TWIK-related acid-sensitive K⫹ channel” (TASK), to name a few. Functional channels are composed of two subunits, each comprising four transmembrane segments and two pore-forming (P) domains. The structures of K2P1.1 and K2P4.1 were solved in 2012 (54, 257) (FIGURE 4B). Assembly with traditional K⫹ channel -subunits leading to functional changes has been proposed (217). The channels are so-called leak or background channels that conduct currents at all phases in the cardiac action potential (FIGURE 2). This conductance ensures the stabilization of the resting membrane potential (Em) towards the equilibrium potential of potassium. Open K2P channels will probably both antagonize early afterdepolarizations and play a role in tuning NaV channel availability (16). The K2P channels have been described in a variety of species including human, rat, mouse, guinea pig, and dog (reviewed in Ref. 166). In human hearts, mRNA expression has been shown for K2P1.1 (TWIK-1/KCNK1), K2P3.1 (TASK-1/ KCNK3), K2P4.1 (TRAAK/KCNK4), K2P5.1 (TASK-2/ KCNK5), K2P15.1 (TASK-5/KCNK15), and K2P17.1 K2P3.1 was originally described by Duprat et al. (109) in 1997. Functional characterization revealed a K⫹-selective, voltage-independent channel (FIGURE 4C). Furthermore, the channels were highly sensitive to changes in external pH near physiological values (109) and were inhibited by the endocannabinoid anandamide (270). Meanwhile, more selective tool compounds have been developed (356, 407). The aromatic carbonamide A293 (Sanofi-Aventis) was shown to inhibit K2P3.1 currents at low concentrations (IC50 222 ⫾ 38 nM in Xenopus laevis oocytes). Furthermore, A293 increased the action potential duration in rat ventricular myocytes, although under nonphysiological conditions (356). It should be mentioned that a controversy exists regarding the contribution of K2P3.1 in rat atria and ventricles. Kim et al. (215) observed a similar expression pattern in the chambers using Northern blot analysis, whereas Liu and Saint (253) reported a more prominent expression in atria compared with ventricles employing quantitative PCR. Evidence for protein expression is sparse due to the lack of specific antibodies in the field (107). Most of the studies on cardiac K2P3.1 function have used mouse cardiomyocytes. In mice, the channel shows a predominantly ventricular expression (107). Taking advantage of a K2P3.1 mouse knockout model, Donner et al. (107) showed that K2P3.1(⫺/⫺) mice display prolongation of the rate-corrected QT interval and reduced heart rat variability. Assessment of time and frequency domains as well as baroreceptor reflex revealed sympathovagal imbalances (107) increasing the impact of sympathetic stimulation in K2P3.1(⫺/⫺) mice. In a follow-up study, the same group showed that the key electrophysiological parameters such as electrical conduction and refractoriness were unchanged in K2P3.1(⫺/⫺) mice, yet the heart rate response after premature ventricular contractions was significantly abolished. The authors suggested that the baroreceptor reflex in K2P3.1(⫺/⫺) is diminished either due to sympathetic predominance or an impaired cardiac parasympathetic activity (342). In a parallel study employing A293, Decher et al. (95) also reported that ITASK plays a role in modulation of cardiac action potential duration and that genetic silencing of K2P3.1 leads to broadened QRS complexes in a mouse knockout model (95). The results obtained from the K2P3.1 knockout mouse model only partially translate to human physiology given the predominant atrial expression in humans (131, 250). Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org 617 Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 2. Two-pore domain K⫹ channels (TASK-4/KCNK17) (21, 94, 131, 325, 327). Most experimental and functional evidence in human heart is reported for K2P1.1 and K2P3.1, the latter being predominantly expressed in the atrium and the atrioventricular node (117, 131, 250). Hence, K2P3.1 has been added to the list of prominent candidates for developing atria-selective drugs. The following section will give an account of the current knowledge about K2P3.1 function in the heart. SCHMITT, GRUNNET, AND OLESEN A K2P10.1 (NP_612190) K2P2.1 (NP_001017424) K2P4.1 (NP_201567) K2P5.1 (NP_003731) K2P17.1 (NP_113648) K2P7.1 (NP_203133) K2P1.1 (NP_002236) K2P6.1 (NP_004814) K2P12.1 (NP_071338) K2P13.1 (NP_071337) K2P9.1 (NP_057685) K2P15.1 (NP_071753) K2P16.1 (NP_001128577) K2P18.1 (NP_862823.1) 180 160 140 120 100 B 80 60 40 20 0 C K2P3.1 mock 200pA K2P1.1 100 ms 100 mV -80 mV -120 mV FIGURE 4. Overview of K2P channels. A: phylogenetic tree of K2P channel proteins. Alignment is as in Figure 3. In red are channels for which mRNA expression has been reported in the human heart (see text for details). B: ribbon representation of the recently resolved K2P1.1 structure based on Protein Data Bank ID 3UKM (283). Transmembrane segments of one subunit are colored (TM1, blue; TM2, purple; TM3, pink; TM4, red). The so-called C-helix located just below the lipid surface is labeled in green; the lipid bilayer is indicated by the black bar. (Picture was generated with Protean 3D, DNASTAR program package.) C: representative patch-clamp recordings of CHO-K1 cells transiently expressing K2P3.1, or mock-transfected cells. Current was recorded during 500-ms voltage-clamp pulses from ⫺120 to 100 mV in 20-mV steps from a holding potential of ⫺80 mV. 618 Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 K2P3.1 (NP_002237) CARDIAC POTASSIUM CHANNELS AND ARRHYTHMIA Limberg et al. (250) investigated the role of K2P3.1 channels in human atria. They showed that a current carried by K2P3.1, ITASK, accounts for ⬃40% of background current in human atrial myocytes isolated from healthy humans. Furthermore, applying a mathematical model adapted to atrial action potential, the authors showed that changes in ITASK may alter the duration of the atrial action potential (250). Despite the apparent role of K2P channels for setting the resting membrane potential and participating in repolarization, only a few reports exist on disease-associated mutations in these channels. Lafrenière et al. (229) reported a dominant-negative mutation in K2P18.1 (TRESK/KCNK18) linked to a form of familial migraine. An abstract written in German alludes to a gain-of-function K2P17.1 (TASK-4/KCNK17) variant associated with conduction defects (127). Of note, Barbuti et al. (27) described a role for K2P3.1 channels in the arrhythmic effects of inflammatory phospholipid platelet-activating factor (PAF) in mice. PAF is released upon reperfusion after ischemic events and triggers arrhythmia (262). Barbuti et al. (27) showed that PAF action induced spontaneous beats, EADs, and prolonged depolarization similar to previous observations in canine cardiomyocytes. These effects involved PKC-dependent block of K2P3.1 (27). The same group recently reported an association between K2P3.1 inhibition with atrial fibrillation in a canine model of peri-operative AF (113). However, the study did not irrevocably show whether K2P3.1 inhibition contributed to arrhythmogenesis or whether it was secondary to AF induction in this specific animal model (173). In addition, it should be kept in mind that K2P3.1 function has also been related to other pathophysiological conditions such as ischemia, cancer development, inflammation and epilepsy (reviewed in Ref. 44). Hence, therapeutic value has to be carefully considered given the wide distribution of these channels in tissues other than the heart. Although K2P3.1 channels are attractive candidates for the development of atrial-specific drugs, further studies are warranted to elucidate whether K2P3.1 inhibitors are antiarrhythmic or proarrhythmic. C. Striking the Wrong Note: Genetic Variation Causing Arrhythmia Several ventricular and supraventricular arrhythmias such as long and short QT syndromes (LQTS, SQTS), Brugada syndrome (BrS), Andersen’s syndrome, and AF have been associated with impaired potassium channel function (reviewed in Ref. 311). Disease-associated mutations in the genes encoding the repolarizing potassium channels of the heart have been instrumental in understanding the channels’ physiological function and their role in pathophysiological conditions. TABLE 2 gives an overview of potassium channel subunits associated with arrhythmia. In the case of ventricular arrhythmias, loss-of-function of potassium conductance leads to action potential prolongation and syndromes such as the LQTS. Vice versa, gain-of-function results in shortening action potential duration and SQTS (174). Supraventricular arrhythmias such as AF show a more complex pattern as both loss-offunction and gain-of-function mutations in a single potassium channel have been described (e.g., Ref. 79), as well as mutations showing a mixed phenotype of AF and QT prolongation (265). AF as well as LQTS are generally considered to be driven by triggered activity and electrical reentry. In both conditions, action potential prolongation tends to increase the triggered activity, whereas action potential shortening, as it is observed in AF and in SQTS, promotes reentry. In general, the heart rate corrected QT interval (QTc) is considered to be a measure of ventricular repolarization. Its prolongation has been associated with an increased risk of sudden cardiac death (494), although its predictive value for ventricular arrhythmia is debated and the concept of TRIaD (triangulation, reverse use depen- Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org 619 Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 The latter observation supports the notion that K2P3.1 may be a valuable target candidate for the development of drugs against atrial fibrillation. Inhibition of K2P3.1 is speculated to prolong action potential duration and effective refractory period and to suppress atrial arrhythmias. Recently, several antiarrhythmic drugs have been applied to K2P3.1 channels. Carvedilol inhibited heterologously expressed K2P3.1 with IC50 ⫽ 0.83 M in mammalian Chinese hamster ovary (CHO) cells (405). The class III antiarrhythmic drug amiodarone was shown to inhibit K2P3.1 channels in Xenopus laevis oocytes (IC50 ⫽ 0.4 M) without changing rectification properties (139). Also dronedarone inhibits K2P3.1 channels in both Xenopus laevis oocytes (IC50 ⫽ 18.7 M) and CHO cells (IC50 ⫽ 5.2 M) (379). A more specific drug, A1899 (Aventis), was recently reported. The compound inhibited K2P3.1 as an open-channel blocker with an IC50 ⫽ 7 nM in CHO cells, and bound to the closely related K2P9.1 with a 10-fold lower affinity. Furthermore, A1899 did not affect a large panel of other channels expressed in human cardiomyocytes (407). The recent publication of the human K2P1.1 crystal structure could enable the development of specific drugs (257). Very recently, impaired K2P3.1 function was postulated as an arrhythmogenic substrate (247). Transient knockdown of K2P3.1 expression affected atrial size in a zebrafish model. Furthermore, reduced K2P3.1 current prolonged atrial action potential duration in cardiac action potential modeling, which was potentiated by reciprocal changes in activity of other ion channel currents (247). SCHMITT, GRUNNET, AND OLESEN Table 2. Potassium channel mutations associated with arrhythmia Protein/Subunit Gene Current Effect of Mutation Type of Arrhythmia Reference Nos. Familial atrial fibrillation Early-onset lone atrial fibrillation Early-onset lone atrial fibrillation Early-onset lone atrial fibrillation Long QT syndrome Short QT syndrome Long QT syndrome Short QT syndrome Long QT syndrome Andersen Syndrome Short QT syndrome Atrial fibrillation Atrial fibrillation Long QT syndrome Conduction defect 298, 322 79 79 321 298, 377 55, 298 298, 442 34, 298 298, 348 298, 348 298, 355 467 64 478 127 Long QT syndrome Early-onset lone atrial fibrillation Long QT syndrome Atrial fibrillation Atrial fibrillation Long QT syndrome Brugada syndrome Atrial fibrillation Brugada syndrome 298, 307 319 2, 298 477 264, 489 316 96, 296 360 317 ␣-Subunits KCNA5 IKur KV4.3 KV11.1 KCND3 KCNH2 Ito IKr KV7.1 KCNQ1 IKs Kir2.1 KCNJ2 IK1 Kir3.4 KCNJ5 IKAch K2P17.1 KCNK17 ITASK Loss of function Gain of function Gain of function Loss of function Gain of function Loss of function Gain of function Loss of function Loss of function Gain of function Gain of function Loss of function Loss of function Gain of function -Subunits KCNE1 KCNE1 KCNE2 KCNE2 KCNE3 KCNE3 KCNE5 KCNE5 IKs IKs IKr IKs IKr/Ito IKs Ito IKs Ito Loss of function Gain of function Loss of function Gain of function Gain of function Loss of function Gain of function Gain of function Gain of function If more than two reports exist, the original reference is provided together with a link to a mutation database or relevant review. dence, instability and dispersion) has been suggested (184, 186, 187). However, there is increasing evidence that prolongation of the mean QT interval is associated with AF (330). A recent study including digital ECGs from more than 300,000 persons confirmed that the QTc interval length is a predictive parameter for the susceptibility of AF (308). Of note, both shortened and in particular prolonged QTc interval duration are risk factors for AF (308). This association was strongest for lone AF cases, a condition in which other predisposing diseases such as hypertension, diabetes, ischemic, and/or valvular heart diseases are absent (66, 67). The study indicates that QTc prolongation is an inherent characteristic of an individual’s cardiac electrophysiology and that prolonged QTc can predispose to AF (308). Vice versa, Johnson et al. (199) have previously shown a prevalence of early-onset atrial fibrillation in congenital LQTS. Hence, ventricular and atrial arrhythmia phenotypes seem to overlap, although knowledge about the mechanisms on the cellular and molecular levels is only slowly accumulating. 620 Major challenges in elucidating these mechanisms are the often observed 1) low penetrance in families and 2) variable expressivity in mutation carriers. As illustrated in FIGURE 5, penetrance describes the extent to which all mutation carriers within a family develop the clinical symptoms of the respective disease. Variable expressivity designates the degree to which individuals with the same genotype express a certain phenotype. Congenital LQTS is a paradigm for such an arrhythmia with incomplete penetrance and variable expressivity (reviewed in Ref. 141). It has been recognized that additional factors contribute to the manifestation of the arrhythmia and the observable symptoms. The impact of factors such as gender, age, seasonal changes, circadian rhythm, and triggers such as auditory stimuli or exercise, or physiological challenges such as hypokalemia have been reported (42, 89, 152, 165, 227, 290, 414, 486). Beyond environmental modifiers, the presence of additional genetic modifiers is well-recognized. Both compound heterozygosity and digenic heterozygosity have been described (381, 453). A recent study has shown that LQTS patients with multiple Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 KV1.5 CARDIAC POTASSIUM CHANNELS AND ARRHYTHMIA ion channel function and hence cardiac rhythm. Yet, these nonsynonymous variants of potassium channel genes also present in the healthy population may act as modulators of the cardiac action potential and as a “second hit” when the individual faces physiological challenges (reviewed in Ref. 141). A B Noteworthy, the increasing availability of genomic data is challenging the concept of channelopathies as monogenic disease as well as aiding understanding of the basis of arrhythmia. Analyses of continuously updated exome data provided by the NHLI GO Exome Sequencing Project (ESP) currently holding data from 6500 individuals (119a) revealed that many previously disease-associated mutations are in fact variants that occur frequently in the general population (14, 363). Hence, these variants might not be the underlying cause of the disease. C Genotype-negative ECG phenotype Genotype-positive ECG + cardiac event FIGURE 5. Complexity of genetics traits. A: complete penetrance: the genotype of an individual correlates completely with the diagnostic and clinical phenotype. B: incomplete penetrance: not all genotype-positive individuals display ECG and clinical phenotypes. C: incomplete penetrance combined with variable expressivity: genotypepositive individuals display ECG, but not clinical phenotype. [From Guidicessi and Ackerman (141). Copyright 2013, with permission from Elsevier.] mutations in the known LQT genes had a higher rate of life-threatening cardiac events (291). Yet, a general picture emerges that shifts the focus from monogenic inheritance, where very rare variants with a frequency ⬍0.1% in the general population are considered disease causing, to oligogenic (0.1% to ⬍ 5%) or polygenic (common variants ⬎5%) inheritance patterns. It seems that the more frequent the genetic variant, the lower its effect on Solid conclusions regarding a causative link between genomic variant and disease require a combination of 1) detailed analysis of frequency in the general population, 2) algorithms to predict the pathogenic effects of the variant (reviewed in Ref. 420), 3) verification of findings in independent cohorts, 4) often laborious and time-consuming experimental knowledge using suitable in vitro assays, and 5) evaluation of cosegregation in families. The latter is often hampered by small family sizes, lack of Mendelian inheritance, lack of penetrance, and/or variable expressivity. A wealth of publications on variants and mutations, functional data, and possible new players important for cardiac rhythm are continuously being released, which challenges the attempts to see the larger picture of the disease-causing processes. For a decade, Napolitano and Wilson (298) have put tremendous effort into maintaining a large annotated database listing mutations and common variants associated with cardiac arrhythmia. Owing to the complexity of the genetic traits, the availability of genomic data, and the ever improving systems for functional characterization, it is likely that the field will benefit from continuous efforts to collect all available data, including negative findings, in such an arrhythmia database maintained by the scientific community. Such information would be valuable as a guide Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org 621 Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 Several recent genome-wide association studies (GWAS) followed the hypothesis that traits such as heart rate or QTc interval used as markers for arrhythmia are associated with single-nucleotide polymorphisms (SNPs) (reviewed in Refs. 282, 394). The analysis of many common genetic variants in different individuals led to the identification of a number of candidate genes such as KCNN3 encoding KCa2.3 (SK3) channels (118). Although this association has been supported by other studies (70, 320), no disease-causing mutations in the exons or splice sites of KCNN3 have been reported to date. SCHMITT, GRUNNET, AND OLESEN to understanding the physiological/pathophysiological mechanisms of arrhythmia, as well as for risk stratification of affected family members. D. Tuning the Instruments 1. Posttranscriptional mechanisms Alternative splicing of mRNA transcripts has been described for most of the repolarizing potassium channels of the heart. Accordingly, several isoforms with different functional phenotypes exist (22, 200, 223, 327, 402, 404, 460, 469, 499). The alternative splicing increases the redundancy of the channels, but also helps to diversify the function of the multimeric proteins. Some isoforms are highly expressed in the heart but, surprisingly, exert dominantnegative effect on channel function when coexpressed in heterologous expression systems (201, 228, 287). Differential expression of KV7.1 isoforms KV7.1-iso1 and KV7.1iso2 across the human ventricular wall has been suggested to contribute to action potential heterogeneity (338). Whereas the total expression level in the endocardial, midmyocardial, and epicardial layers was similar, the proportion of the dominant-negative KV7.1-iso2 was higher in midmyocardial tissue. Mimicking the experimentally determined ratios in heterologous expression systems indicated a drastically reduced midmyocardial IKs compared with that in the epicardium. (338). Consequently, endo- and epicardial myocytes are reported to have shorter action potentials (57, 251). Recently, Lee et al. (234) determined the effect of amiloride on the ratio of these KV7.1 isoforms in canine endocardial, midmyocardial, and epicardial ventricular myocytes. Whereas the drug led to decreased expression levels of KV7.1-iso1 in endocardial and epicardial layers, KV7.1-iso1 was increased in midmyocardium. The authors further investigated the impact of these changes on action potential duration using simulations of human ventricular action potentials. Their results supported the earlier find- 622 Assembly of different KV11.1 isoforms in the native channel complex influences the kinetics (233) and pharmacological sensitivity (231) and has been implicated in arrhythmia (372, 422). Accordingly, a number of splice-site mutations have been associated with arrhythmia (298). An additional regulatory mechanism, nonsense-mediated mRNA decay (NMD), has been described for KV11.1 channels. NMD is an RNA surveillance mechanism that selectively degrades mRNA transcripts that have in-frame premature termination codons. Nonsense mutations have been implicated in human disease (289), and NMD is thought to serve as a “cellular vacuum cleaner” that protects the cell from the potentially harmful effects of truncated proteins by eliminating mRNAs with premature termination codons. NMD occurs when translation terminates 50 –55 nucleotides upstream of the 3’-most exon-exon junction (33). Gong and co-workers investigated LQT2 nonsense mutations that showed a decrease in mutant mRNAs by NMD, thereby altering the amount of mRNA available for subsequent KV11.1 protein generation. It was suggested that the degradation of premature termination codons containing mRNA transcripts by NMD represents a new class of LQT2 pathogenic mechanism (148, 149, 410, 487). Yet of all the channel proteins involved in cardiac excitability described above, this mechanism has been exclusively described for KV11.1 channels. Traditionally, the field has focused on the protein encoding sequence of DNA and paid only scant attention to 5’- or 3’-untranslated regions (UTR). This changed with the discovery of so-called microRNAs. These small noncoding RNAs are part of the epigenetic control of gene expression that also encompasses DNA methylation and histone modifications (143). The binding of miRNAs to the 3’-UTR of their targets can lead to degradation of target mRNAs or repression of target mRNA translation. A number of cardiovascular-specific miRNAs have been described (reviewed in Ref. 45). A very interesting feature is the fact that miRNAs may regulate multiple targets simultaneously, which allows for complex and cooperative regulation of several channel proteins whose functions are tightly connected. Also, several miRNAs can bind to the same target, which increases the level of regulatory control. Although the field is only emerging, several miRNAs have been linked to the regulation of the cardiac repolarizing potassium channels (216). Hence, changed miRNAs levels in disease conditions may have an impact on cardiac excitability. Examples are the downregulation of miR-1 in atrial fibrillation leading to increased IK1 currents (140) or the upregulation of miR-301a in a diabetic mouse model leading to Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 The delicate act of keeping the heart beat stable under all conditions is quite sensitive to changes in the function of the key ion channels. The expression and activity of the channels may vary significantly as a consequence of posttranscriptional and epigenetic changes. Furthermore, the channel function is also drastically influenced by the proteins’ exact subcellular localization within cardiac myocytes; the complexes they form with accessory subunits, kinases, and phosphatases; and posttranslational modifications. As elaborated above, channel function can change due to mutations, yet also due to electrical remodeling and presence of drugs. Below we will give a short account of some of these important mechanisms that influence the function of the channel on its way from the gene in the nucleus to the surface-expressed protein, and their significance for cardiac repolarization. FIGURE 6 gives a summarized picture of the outlined regulatory mechanisms. ings that transmural differences in IKs density might be explained by the ratio of the two isoforms (234, 338) and indicated that amiloride may lead to a reduction of the transmural IKs gradient (234). CARDIAC POTASSIUM CHANNELS AND ARRHYTHMIA K+ K+ Kinase K+ Phosphatase P Ub Vesicle Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 pH Lysosome Ub Ub Ub Ub Proteasome Golgi ER Vesicles Translation repression miRNA Nucleus RISC Alternatice splicing RISC DNA pre-miRNA miRNA Mutually exclusive exons NMD STOP AUG RNA Exon skipping Alternative 5’ donor sites Cap Ex1 Ex2 Ex3 Ex4 (A)n PolyA Distance >55 nucleotides Potential NMD Targets ≤55 nucleotides Non NMD Targets Alternative 3’ donor sites Intron retention FIGURE 6. Postranscriptional and posttranslational regulatory mechanisms. After transcription in the nucleus, mRNA is processed, alternatively spliced, and translated into protein, unless degraded via, e.g., nonsense-mediated mRNA decay (NMD) or miRNA mediated translation repression. Proteins are folded in the ER. Misfolded proteins are degraded in the proteasome (indicated by the dashed line), whereas correctly assembled proteins are processed in the Golgi apparatus, where glycosylation may occur. Mature channel complexes are transported in vesicles to the plasma membrane. Targeting signals in the protein may guide the membrane protein to a specific location within the cardiomyocyte, e.g., t tubules or intercalated discs. The potassium ␣-subunit can interact with accessory subunits and undergo additional posttranslational modifications such as phosphorylation (P) and ubiquitination (Ub). Ubiquitinated proteins are endocytosed and degraded in the proteasome (via lysosomes). Alternatively, the channel can be recycled to the membrane in recycling vesicles (not shown). Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org 623 SCHMITT, GRUNNET, AND OLESEN decreased Ito (332). An overview of known miRNAs affecting cardiac potassium currents is given in TABLE 3. Further research exploring these outline mechanisms will hopefully shed additional light on the determinants of low penetrance and variable expressivity in arrhythmia research. 2. Posttranslational regulation of potassium channels in protein complexes Potassium channels are often pictured as isolated membrane proteins, but it is well recognized that they are part of Of all the cardiac ion channel complexes, the IKs channel has probably received the most attention due to its regulation by the KCNE family of -subunits. The interaction with KCNE1 (29, 376) leads to dramatic changes in biophysical properties such as a major increase of macroscopic current, slowed activation, and shift of the voltage dependence of activation to more depolarized potential (reviewed in Ref. 464). The exact mode of interaction between these subunits and their stoichiometry has been intensively studied and is still a matter of debate. The interaction seems to be dynamic with variable stoichiometry and interaction sites (reviewed in Ref. 464). In addition to KCNE1, the ␣-subunit is able to interact with all five members of the KCNE channel family in heterologous expression systems (197). The relative expression of KCNE genes in human hearts is KCNE4 ⫽ KCNE1 ⬎ KCNE3 ⬎ KCNE2 ⬎ KCNE5 (35). Despite this, the only functionally important interaction is probably KV7.1/KCNE1, as KCNE4 and KCNE5 significantly right-shift the activation curve for the channel complex, rendering them nonfunctional under normal physiological conditions (15, 158, 160). The interaction with KCNE2 and KCNE3 seems to be most important in non-cardiac tissue (35, 195), although mutations in KCNE2 and KCNE3 genes have been associated with cardiac arrhythmia (see below), pointing to a functional role of these subunits in the heart (96, 116, 264, 316, 477). Table 3. microRNAs regulating cardiac potassium channels miRNA Target Gene/Protein miR-1 Ito KCND2/KV4.2 miR-26 IK1 IK1 KCNJ2/Kir2.1 KCNJ2/Kir2.1 miR-133 Ito ND IKr KCNH2/KV11.1 IK1 Ito KCNJ2/Kir2.1 KCND2/KV4.2 miR-212 miR-301a Comment Reference Nos. Deletion of miR-1 leads to widened QRS complex, effect mediated via transcription factor Irx5 (mouse model) Reduced miR-1 level in atrial fibrillation leads to increased IK1 (human) Reduced miR-1 level in atrial fibrillation leads to increased IK1 (human, and canine and mouse models) Increased miR133 level leads to decreased Ito possibly by an indirect mechanism involving KChIP2 (mouse model) Increased miR133 level leads to decreased KV11.1 protein levels and reduced IKr (guinea pig model) Downregulates Kir2.1 and IK1 (HeLa cells) Increased miR-301a levels lead to decreased KV4.2 protein level, direct binding to 3’UTR (diabetic mouse model, db/db) 497 ND, not determined. 624 Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org 140 267 278 387 144 332 Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 Adding another level of complexity, Amin et al. (12) reported that common variants in the 3’-UTR of the KCNQ1 gene encoding KV7.1 modified disease severity in LQTS patients in an allele-specific manner. As KV7.1 channels are composed of tetramers, in a heterozygous mutation carrier channels will be formed from two healthy and two mutant subunits, provided that wild-type and mutant alleles are expressed equally well. The authors found several SNPs in the 3’-UTR of the KCNQ1 gene. With the assumption that the SNPs exert no effect on expression, the balance between normal and mutant subunits within each channel should be equal. However, the authors suggested a model in which the subunits carrying the disease-modifying SNPs were suppressed, which would affect the balance between wild-type and mutant channel subunits. Such suppression could be mediated by miRNAs that bind to consensus sites in the 3’-UTR. Depending on which nucleotide sequence favors the binding of miRNAs, then the balance of wild-type and mutant subunits relies on whether the “suppressive” SNP variants reside on the normal or mutant allele. Accordingly, an increased expression of the mutant allele would enhance the disease phenotype of the patient, while an increased expression of the wild-type allele could possibly result in an asymptomatic phenotype (12). large complexes. They form multimolecular complexes with each other as well as with accessory subunits, anchoring proteins, and regulatory kinases and phosphatases. The association with non-pore-forming proteins, so-called -subunits, helps to regulate and modify the function of the ␣-subunit, likely in a temporal and spatial manner. A number of excellent recent review articles have covered the composition and regulation by accessory subunits for the protein complexes underlying Ito, IKur, IKAch, IKr, and IKs currents (268, 311, 359, 433, 464). CARDIAC POTASSIUM CHANNELS AND ARRHYTHMIA In addition, these multiprotein complexes link membrane proteins to intracellular proteins and contain components of the cytoskeleton and extracellular matrix proteins. Furthermore, cardiac myocytes are polarized cells with most ion channels localized at specific subdomains such as the t-tubular invaginations or intercalated disks. Hence, highly regulated trafficking and targeting mechanisms are crucial to achieving these specific locations. The mechanism and the current view on where the potassium channel complexes are located within the surface membrane of cardiomyocytes are reviewed in Reference 26. Protein activity is not only controlled by the rates of protein biosynthesis and protein degradation, but also by specific and selective covalent processing, i.e., posttranslational modification (PTM) such as phosphorylation, which modulates molecular interactions, protein localization, and stability. TABLE 4 lists the current knowledge on PTMs for the major repolarizing currents of the heart. These modifications may provide an important feedback mechanism for regulating the number of functional complexes upon external stimuli from, e.g., hormones and the autonomic nervous system, which will significantly influence cardiac signaling. The greater our understanding of the signaling pathways, the more complex the picture may become. Several studies have shown that common genetic variants in the components that modulate potassium channel function such as G protein-coupled receptors or kinases may have an influence on cardiac signaling. SNPs in the genes encoding ␣2 and 1 adrenergic receptors, ADRA2C and ADRB1, respectively, have been linked to the LQTS disease phenotype (reviewed in Ref. 141). Furthermore, a recent study on chronic AF in humans showed a chamberspecific upregulation of 1 adrenergic receptors, which led to dramatic effects on several repolarizing currents, foremost IKs (150). Other regulatory pathways such as the nitric oxide pathway also seem to be linked to cardiac arrhythmia. Arking et al. (19) were the first to establish a link between the age, gender, and heart rate corrected QT-interval with SNPs in the gene encoding nitric oxide synthase 1 adapter protein (NOS1AP, also known as CAPON) (19). The SNP rs10494366 located in the upstream region of the coding region achieved genome-wide significance. This finding has been replicated in several large independent populations of both European ancestry and other ethnicities. In those studies, several additional NOS1AP SNPs were found to be significantly linked to the QTc interval (1, 18, 114, 274, 306, 343, 352). Noteworthy, NOS1AP SNPs reached higher significance compared with SNPs in genes known to be causes of LQTS and SQTS (306, 343). Furthermore, NOS1AP has been linked to sudden cardiac death (205, 326). Analyzing the protein complexes and their regulation in the cellular network will help our understanding of the complex regulation of cardiac excitability. III. MECHANISMS AND ETIOLOGY OF ARRHYTHMIAS Arrhythmia in its broadest sense describes every type of abnormal heart rhythm ranging from gentle transient palpitations to devastating fibrillations. Ultimately, fibrillation in the ventricles can result in sudden cardiac arrest. A subdivision of arrhythmias encompasses bradycardia and tachycardia, representing too slow or too rapid heart rates, respectively. Heart rates are species dependent, and humans are considered bradycardiac when having fewer than 60 bpm and tachycardic with more than 100 bpm. In brady- or tachycardia the individual can either have a regular rhythm triggered by the sinus node (sinus rhythm), or an irregular arrhythmia governed by other mechanisms. In AF, the atrial rhythm is ⬎200 bpm. While not lethal, it is associated with thromboembolic events and an increased risk of cardiac death in a longer perspective (36). This increased morbidity and mortality is the result of the reduced ejection fraction during tachycardia and blood stasis in the fibrillating atria increasing the propensity of thrombosis and stroke (461). Ventricular arrhythmias are significantly more serious as they directly influence the pumping capacity of the heart. A ventricular tachyarrhythmia can be both monomorphic and polymorphic in origin and can eventually develop into fibrillation. In ventricular fibrillation, the electrical activity of the cardiac cells occurs in a completely uncoordinated manner and pumping ceases. From a mechanistic perspective, tachyarrhythmic events are obligate depending on two phenomena: a triggering event for initiation and a substrate for sustainability. In the following, we give a thorough overview of triggers and substrates. This includes an introduction to the spiral wave and Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org 625 Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 In 2012, Milstein et al. (285) used a multipronged approach to show a large macromolecular complex involving both Kir2.1 and NaV1.5 channels. They thereby revealed an extraordinarily tight connection between the IK1 current controlling the resting membrane potential and INa, whose availability, effect on cell excitability, action potential duration, and velocity of impulse propagation are governed by the membrane potential set by IK1 (285). Noteworthy, the postsynaptic density, discs large and zonula occludens-1 (PDZ) domain protein SAP97 is a component of this macromolecular complex (285). The same anchoring protein has previously been shown to interact with KV1.5 in both heterologous expression systems (292) and cardiomyocytes, where it seems to stabilize the channels in the plasma membrane (72). This opens up for the possibility of even larger macromolecular complexes also involving IKur channels. SCHMITT, GRUNNET, AND OLESEN Table 4. Posttranslational modifications of cardiac potassium channels ␣-Subunit KV1.5 KV4.3 KV11.1 Kir2.1 IKur Ito IKs IKr IK1 Kir2.2 IK1 Kir3.1 IKAch Kir3.4 IKAch Kir6.2 IKATP K2P3.1 Ileak Modification Enzyme Acetylation Glycosylation Palmitoylation Sumoylation Nitrosylation Phosphorylation Phosphorylation Phosphorylation Phosphorylation Phosphorylation Phosphorylation Phosphorylation Phosphorylation Phosphorylation Src EGFR kinase Src p90RSK PKC CaMKII CaMKII PKA PKC Dephophorylation Ubiquitination Deubiquitination PP1 Nedd4-2 USP2 Glycosylation Glycosylation Phosphorylation Phosphorylation Ubiquitination Acetylation Nitrosylation Phosphorylation Phosphorylation Phosphorylation Phosphorylation Glycosylation Glycosylation Phosphorylation Phosphorylation Phosphorylation Phosphorylation Phosphorylation Phosphorylation Phosphorylation Phosphorylation Phosphorylation Ubiquitination Phosphorylation Phosphorylation Phosphorylation PAT Ubc9 Amino Acid Position(s) C604 N290 C593 K221, K536 C331, C346 n.d. Y136 Y108 S516, S550 T503 S550 S27 S409, T513, S464, S577 (S27) P660, Y662 (P660, Y662) Src Nedd4-2 N598, N629 N598 S283, S890, T895, S1137 Y475 P1075, Y1077 PKA PTK PKA PKC C76 S425 Y242 S425 S64A, T353A PKA N119 PKA PKC PKC CaMKI CaMKII CaMKII PKC PKA PKC PKC Src RhoK S185 S191 S372 T180 T381 S336 Species h h r h h h h h h h r h h h h h,gp h Native ⫹ ⫹ ⫹ ⫹ ⫹ — — — — — (ventricle) (atria) (atria) — (ventricle) (heart) — — — 276 196 225 — — — 341 147 419 — ⫹ (gp heart) — ⫹ (atria) — — ⫹ (ventricles) ⫹ (atria) ⫹ (atria) — ⫹ (atria) ⫹ (atria) — ⫹ (atria) ⫹ (atria) ⫹ (atria) — — — — — — — Only direct posttranslational modifications that have been reported in either mammalian cell systems or cardiac tissue are listed. References are given for the first report on the respective modification. If known, the amino acid position referring to full-length protein and enzyme are given. Note that some regulatory pathways act via accessory subunits not included in this list. CaMK, calmodulin-dependent kinase; EGFR, epidermal growth factor receptor; gp, guinea pig; h, human; Nedd4, neuronal precursor cell-expressed developmentally downregulated 4; PAT, palmitoyl acyltransferase; PKA, protein kinase A; PKC, protein kinase C; PP1, protein phosphatase 1; PTK, phosphotyrosine kinase; p90RSK, p90 ribosomal S6 kinase; r, rat; RhoK, Rho kinase; USP2, ubiquitin-specific protease 2. 626 493 382 198 38 313 182 495 495 261 349 80 385 276 208 — ⫹ (gp ventricles) — h h h h h,gp m h r h r h m h r r h r r r h h m r m Reference Nos. Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org 490 5 429 146 459 491 236 214 212 329 281 281 272 281 281 281 272 32 249 417 43 294 386 Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 KV7.1 Current CARDIAC POTASSIUM CHANNELS AND ARRHYTHMIA leading circle theories explaining reentry-based arrhythmias to understand the role of the repolarizing currents in these arrhythmias. A. Triggers of Arrhythmias The initiation of an arrhythmia requires a triggering event. Abnormal focal automaticity constitutes a typical trigger. Several areas of the heart have the intrinsic capability for automaticity or pacemaker activity, but under normal circumstances the sinoatrial node has the fastest pacemaker rate and will therefore be the area that determines the heart rate. Impulse generation in areas outside the sinoatrial or atrioventricular nodes is called abnormal automaticity, and the abnormal pacing site is called an ectopic focus. Several factors can increase the risk of abnormal automaticity, such as increased sympathetic drive, acute myocardial ischemia, or electrical remodeling leading to changed ion channel distribution (51, 358). Areas around the left and right pulmonary veins are especially sensitive to triggering events (167). The increased likelihood of focal (nonreentrant) arrhythmias around pulmonary veins is explained by several factors: specialized cells with pacemaker activity are located here, and the resting membrane potential is more depolarized as a consequence of reduced IK1 current (111, 238). Abnormal focal activity can also be initiated by early afterdepolarizations (EADs), which are inappropriate secondary depolarizations taking place during the normal repolarization phase of the cardiac action potential (phase 3) (FIGURE 7). EADs are primarily due to reactivation of the voltage-gated CaV1.2 channels (L-type Ca2⫹ channels). These channels are activated during the initial depolarization of the action potential (phase 0) and will subsequently undergo inactivation in a time-, voltage-, and Ca2⫹-dependent manner. The inacti- Accelerated normal automaticity Another source of abnormal focal activity is delayed afterdepolarizations (DADs) (FIGURE 7). A DAD describes inappropriate depolarizations in the time interval from complete repolarization of an action potential to the upstroke of a subsequent action potential, i.e., during the diastolic interval or phase 4. The mechanistic understanding of DADs relies on inappropriate activity of the Na⫹/Ca2⫹ exchanger. This cotransporter has a stoichiometry of 3 Na⫹ exchanged for 1 Ca2⫹, implying electrogenic transport. The exchanger is the main exit pathway for Ca2⫹ having entered the car- EAD DAD 0 mV 250 ms -80 mV Normal atrial AP Arrythmic APs FIGURE 7. Triggers for arrhythmia: accelerated normal automaticity (left), delayed afterdepolarizations (DADs) (middle), and early afterdepolarizations (EADs). For details, see text. [Modified from Iwasaki et al. (190) with permission from Lippincott Williams and Wilkins/Wolters Kluwer Health.] Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org 627 Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 vation is released by the hyperpolarization of the membrane potential and reduction in [Ca2⫹]i in phase 3. If release from inactivation takes place while the membrane potential is still sufficiently depolarized to allow for activation of the CaV1.2 channels, the result is inappropriate reopening that will prompt an EAD (392, 474, 488). The critical time interval during repolarization that allows for reactivation of the CaV1.2 channels is recognized as the “vulnerable window.” This normally lies from 30 to 90% of full repolarization of the action potential (APD30-APD90). EADs typically originate in ventricular tissue zones with extended action potentials, such as the Purkinje system, but can also be observed in atrial tissue as triggers for atrial fibrillation (58, 378, 473). It is worth mentioning that it is not simply the action potential duration per se that governs whether an EAD might commence. The exact morphology of the action potential in the repolarizing phase 3 is probably more important (184). Delayed repolarization seen as a prolongation of the APD30-APD90 interval will increase triangulation of action potentials and thereby also the time interval for the vulnerable window. This augments the probability for EADs. A typical situation for EAD-triggered arrhythmias in both atria and ventricles is the LQTS (199). SCHMITT, GRUNNET, AND OLESEN diomyocyte through L-type Ca2⫹ channels. The Na⫹/Ca2⫹ exchanger removes one Ca2⫹ in exchange for the inward movement of three Na⫹, which generates a net inward transport of positive ions resulting in a depolarizing current. The effect of this current can normally be counteracted by the large resting K⫹ conductances. In situations with excessive intracellular Ca2⫹ load, this electrogenic exchange generates a depolarization recognized as a DAD (183, 295, 434, 503). Intracellular Ca2⫹ overload can be subsequent to Ca2⫹ leak from mutant ryanodine receptors (RyR) in the sarcoplasmic reticulum, which can result in catecholaminergic polymorphic ventricular tachycardia and concomitant fibrillation (347). Hyperphosphorylation of RyR as a consequence of increased sympathetic drive can also provoke phase 4 Ca2⫹ overload and DADs (106). Congestive heart failure is another source of Ca2⫹ overload and fibrillation (481). unidirectional blocks and the involvement of obstacles or substrates in the development of arrhythmia (FIGURE 8). Almost 50 years ago, a mathematical description and the first modeling of functional reentry was provided, including the notion that the substrate did not necessarily have to be of anatomical origin (286, 303). A historical overview in combination with an excellent description of theories for reentry-based arrhythmias is given by Comtois et al. (81). On the background of these classical experiments, two different theories have been developed to explain reentrybased arrhythmias: the leading circle and spiral wave theories. A brief introduction to both phenomena is useful to facilitate the understanding of the role of ion channels in re-entry mechanisms. Allessi and co-workers (8 –11) were the first to demonstrate and describe functional reentry based on experimental evidence, from which they developed the leading circle model: “The smallest possible pathway in which the impulse can continue to circulate is the circuit in which the stimulating efficacy of the circulating wavefront is just enough to excite the tissue ahead which is still in its relative refractory phase.” The principle is illustrated in FIGURE 9A. In the situation of an unidirectional block (illustrated by the gray square), the leading circle can be defined as the smallest possible circle of continuous propagating electrical activity (illustrated by the heavy black arrow), which is exactly sufficient in length to reach is original point of initiation at the time point when absolute refractoriness has ended and reexcitation can be evoked. It follows that a smaller circle will reach the starting point at a time point when absolute refractoriness still prevails, hence making reexcitation impossible, so the reentry will terminate. Impulses traveling longer circles will have longer turn-around time, and hence the reentry activity will be dominated by the shortest possible circle that therefore receives the name, the leading circle. The length of such a leading circle is called the wavelength (WL), and this is equal to the product of the effective refractory period (ERP) and conduction velocity (CV) as WL ⫽ ERP ⫻ CV. A reduced wavelength will mean that B. Substrates for Reentry-Based Arrhythmias In order for sustained arrhythmias to occur, the triggering event must subsequently initiate a self-sustained episode of action potential propagation. A change in the conduction properties of the cardiac tissue is a prerequisite for such an activity, and that is called a substrate. Substrates can be anatomical obstacles in nature or based on electrophysiological heterogeneity. The shared denominator is that independently of its origin, the substrate will allow for sustained excitation and propagation of action potentials. Such a situation can arise if the tissue is allowed to regain excitability before the propagating wave of electrical signals meets its starting point. Situations that will prompt this are slow propagation of electrical signals or if the signal has to travel a long path to again reach its starting point. The resulting self-sustained and uninterrupted propagation of electrical signals is denoted a reentry-based arrhythmia. A more detailed description of the nature of different substrates is given in the section describing remodeling. The concept of reentry in the context of fibrillation has been known for more than a century, as have the ideas about 1 2 4 3 Normal 628 Re-entry FIGURE 8. Reentry arrhythmias. Propagation of normal action potential (left) and conditions for a reentrant excitation (right). In normal tissue, the electrical signals travel down each branch with equal velocity. If the two branches are connected, the signals will encounter refractory tissue and will not progress. Yet, if one branch exhibits a unidirectional block (indicated in red) (2), the electrical signal will travel down only one branch (1). If the signal enters the common branch at a time where it is not refractory, retrograde signaling (3) and re-excitation can occur resulting in self-sustained propagation (4). Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 1. The leading circle CARDIAC POTASSIUM CHANNELS AND ARRHYTHMIA A B C CELL 2 D 2. The spiral wave APD reduction Passive activity 1. Curvature and/or twisting effect 2. Reduced excitability by effective refractory period FIGURE 9. Substrates for reentry arrhythmias. A: concept of leading circle. The gray block represents a unidirectional block, and the leading circle is defined as the smallest possible circle of continuous self-propagating electrical activity (black arrow). Inside the leading circle, smaller wavelets (small arrows) form and maintain the central core in a refractory state. B: concept of spiral wave. The spiral wave describes a continuous, rapid movement of electrical signals that rotates around a central core. C: the degree of cell-cell coupling will determine the relative impact neighboring cells can exert on each other and thereby also the relative difference in action potential duration (APD) between cells. Black lines represent APD in uncoupled cells that can be of various lengths. If cells increase coupling via gap junctions, the result is a compensatory effect of APD as a consequence of net depolarizing current flowing from cell 2 to cell 1, thereby increasing APD in cell 1 and decreasing APD in cell 2 as represented by the red dotted lines. The degree of cell-cell coupling will thereby determine the effect on relative APD difference between neighboring cells. This can create a current sink that can serve as a passive central sink area in a spiral wave. D: electrotonic effects in a simple spiral wave showing the inner core region. For detailed explanation, see text. [From Comtois et al. (81) by permission of Oxford University Press.] With the development of high-resolution mapping studies and more advanced in silico modeling, it has become evident that reentry explained by spiral waves is more likely to reveal the true nature of reentry-based arrhythmias compared with the leading circle theory. This includes observed properties such as dynamic core behavior, electrogenic source-sink relations, electrotonic interactions, and the predicted response to Na⫹ channel inhibition (class I antiarrhythmics) (81, 91, 286, 457). The concept of a spiral wave has been developed and described by a number of groups (93, 219, 340, 395, 455). In its most simple form, a spiral wave can be described as a continuous, rapid movement of electrical signals that rotates around a central core. An important difference between the leading circle and the spiral wave is that the latter operates with a curved wave front as shown in FIGURE 9. Depolarizing activity originates from a central core, marked by the largest and more heavily dotted circle in FIGURE 9B, and propagates away from the center. The curved depolarization wavefront is depicted by the solid black line and direction of propagation indicated by the black arrows. Since the depolarizing part of the wave is radiating outward, each cell will have to excite more than one cell when moving away from the core. This generates a source to sink mismatch and consequently decreases propagation velocity. The depolarization will naturally be followed by a repolar- Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org 629 Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 CELL 1 more circles can be included within a given tissue area, thereby increasing the risk of reentry-based arrhythmias, so a reduction in WL is considered proarrhythmic. By the same argument, a prolongation of the WL will be antiarrhythmic. This is the basic principle accounting for the efficacy of classic class III antiarrhythmics. These drugs prolong action potential duration by inhibition of the K⫹ channels responsible for phase 3 repolarization (108, 389). ERP is consequently increased, and provided that CV is unchanged, it leads to increased WL and diminished risk of reentry (FIGURE 9) (301). This principle is particularly valid for atrial arrhythmias, whereas in ventricles it has unambiguously been shown that K⫹ channel inhibition can also lead to an increased heterogeneity in the ventricular action potential duration, and this is profibrillatory (17, 82, 353). In the context of AF, it should also be mentioned that structural remodeling leading to atrial enlargement or dilatation is considered proarrhythmic, the reason being that increased tissue area is equivalent to a relative reduction in WL. Physical or electrical atrial remodeling can be caused by valve stenosis, insufficiency, congestive heart failure, or tachycardia and is an important clinical cause of sustained AF (176, 390). The leading circle theory is fascinating in its simplicity and is good for illustrating class III antiarrhythmic efficacy, but from a biophysical perspective, it is probably not the most accurate representation of re-entry based arrhythmias. SCHMITT, GRUNNET, AND OLESEN 3. Implications for modes of drug action An important reality check on the two reentry theories of leading circle versus spiral wave is how they encompass the effects of antiarrhythmic compounds. The leading circle easily explains how the class III compounds inhibiting repolarizing K⫹ currents cause an increased ERP and concomitant increase in WL (WL ⫽ ERP ⫻ CV), which according to the theory should be antiarrhythmic. When explaining the efficacy of K⫹ channel inhibitors on the basis of spiral wave reentry, the situation is more complex, although this model also predicts an antiarrhythmic effect. The exact 630 outcome will, however, depend on which type of K⫹ current is targeted. In general, reduced action potential duration will tend to accelerate and thereby stabilize rotors (31). From this perspective, an antiarrhythmic effect is expected when blocking K⫹ currents, especially when targeting IK1 (373). Other antiarrhythmic effects of K⫹ channel inhibition are explained by an increase in the core area and meandering (31). Yet, in principle, K⫹ current inhibition can also result in formation of additional spiral waves, especially in ventricular tissue. In situations with sufficient excitable tissue, this could lead to fibrillatory activity (134, 362, 412). In attempts to explain the efficacy of class I compounds represented by Na⫹ channel inhibitors, the leading circle theory falls short. Na⫹ channel inhibitors are divided into class Ia, Ib, or Ic according to their effects on Na⫹ channel kinetics being intermediate, fast, or slow, respectively, and on the different effects on action potential morphology (284, 318). All classes will decrease CV, and class Ib will further decrease ERP. Recalling WL ⫽ ERP ⫻ CV, it follows that Na⫹ channel inhibitors will decrease WL via both parameters, and thereby should be proarrhythmic. Although all antiarrhythmics can comprise proarrhythmic properties under certain circumstances, the overall antiarrhythmic effects of class I compounds seem to be in opposition to the leading circle description of reentry. In contrast, antiarrhythmic effects of class I compounds are easily explained from the spiral wave perspective. Na⫹ channel inhibition is expected to increase core size and meandering and to decrease curvature (211, 221). Na⫹ channel inhibition will also diminish the source-to-sink mismatch in the core area of the spiral wave. Altogether these effects should terminate reentry-based arrhythmias (81). It should be noticed that the concepts presented above primarily apply to atrial tissue. In ventricles, the larger muscle mass complicates the nature of reentry, as will be discussed in the section on ventricular fibrillation. Finally, it should be mentioned that during the last 60 years, the spiral wave and leading circle theories have gained most attention for explaining the principles of multiple circuit reentry. There is, however, also more recent experimental evidence to support single reentry circles, mainly in the areas of mitral valves and pulmonary veins as the foundation of especially atrial fibrillation. Depending on the conditions, single reentry, multiple reentry, and ectopic foci can all impact reentry-based arrhythmias to various degrees (301). C. Atrial and Ventricular Fibrillation Having introduced triggers, substrates, and unidirectional block as prerequisites for the development of fibrillation, in this section we give a short introduction to the etiology of atrial and ventricular fibrillation. Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 ization front represented by the solid red line. As a consequence of the curvature of the wave, there is a point or area where the depolarizing and repolarizing fronts meet. This point is termed the phase singularity of the spiral wave and is represented by the inner dashed circle (FIGURE 9B) (81). In the leading circle, the central core is believed to be nonexcitable due to refractoriness obtained from centripetal wavelets originating from the leading circle (represented by black arrows pointing inward in FIGURE 9A). This notion was originally based on measuring very short action potentials with reduced amplitude in the central core (11). Later studies showed that the true nature of the core is better represented by the spiral wave theory describing an excitable but nonexcited core (188, 192, 206). From a biophysical perspective, this is explained by the phase singularity that represents such an excitable area of tissue, which is, however, nonexcited due to the nature and rotation of the spiral wave. Outside this area of phase singularity, there will be a larger area (represented by the outer dashed circle in FIGURE 9B) characterized by electrotonic activity, i.e., a movement of depolarization that can spread from cell to cell but does not have sufficient amplitude to reach the threshold for activation of new action potentials. This area of passive activity will serve as a current sink for surrounding tissue. As a consequence of cell-cell coupling, there is a reduction in action potential duration in the surrounding tissue that serves as a source for the passive central sink area. The principle is illustrated in FIGURE 9, C AND D. These electrotonic properties and the faster repolarization as a consequence of the shortened action potential duration have been demonstrated to be pro-arrhythmic by stabilizing rotor behavior and preventing wave breaks (74, 92). The combined effect of core size, wave curvature, and electrotonic properties will have important impact on the wave tip dynamics. A final important notion about the spiral wave theory is the inclusiveness of meandering, i.e., the turning and winding movement of the wave front through the cardiac tissue. To a large degree, the exact trajectory of a given spiral wave will depend on the wave tip dynamics (123, 207, 458). Generally, a very stable core with a minimum of meandering is considered proarrhythmic (213). This is explained by the reduced chance for moving into refractory tissue and thereby the decreased likelihood of wave-breaks that can terminate the re-entry. CARDIAC POTASSIUM CHANNELS AND ARRHYTHMIA Although not devastating per se, AF can significantly influence life quality, and the disease is associated with a longterm increased risk of death (204). The uncoordinated movements of the atria during AF result in impaired ventricular filling and blood stasis and thrombosis in the atrial appendices. The condition predisposes to heart failure and thromboembolic stroke, respectively (443, 461). The severe consequences of stasis are emphasized by the fact that the risk of stroke is increased by fivefold for patients with AF compared with unaffected individuals. It is further estimated that 15% of all stroke incidences are attributable to AF, a number that increases significantly with age (461). In contrast to AF, ventricular fibrillation (VF) is less common but more serious in nature. VF causes an immediate severe reduction in ventricular ejection fraction due to the fast contractions not being coordinated in the different parts of the ventricle. Systemic need for circulation cannot be met, and if continued, VF will result in sudden cardiac death. According to both the spiral wave and leading circle theories for reentry-based arrhythmias, inhibition of K⫹ channels should prolong ERP and exhibit antiarrhythmic effects. This is, however, not necessarily the reality in ventricular arrhythmia, as demonstrated by the SWORD clinical trial (Survival With Oral D-Sotalol) that was preterminated due to increased mortality (440). The proarrhythmic effect of Dsotalol and other more traditional class III compounds can be explained be several factors. Excessive prolongation of action potentials, especially in situations introducing an extended and slowly decaying phase 3-repolarization, will promote reactivation of voltage-gated Ca2⫹ channels. This is exactly the basis for EADs that can trigger an arrhythmic event such as the polymorphic ventricular tachycardia, also described as torsades de pointes (TdP), which can eventually degenerate into VF (377). An additional mechanistic problem from using class III compounds for the treatment of VF is the reverse use- dependency associated with many of these drugs (108). Reverse use-dependency describes the phenomenon that the efficacy of the drugs in prolonging the action potentials is poor in tachycardia and good in bradycardia, where there is no need for the drug to function and where the risk of EAD-based triggering of arrhythmias is maximal. It should be mentioned that whereas a delayed repolarization can be antiarrhythmic, a slowed depolarizing phase (triangulation) is proarrhythmic, and a common problem with K⫹ channel blocking compounds is that they induce both effects (185). The multichannel blocker amiodarone is a fairly safe compound that prolongs the QT interval (indicating prolonged ventricular action potentials), does not induce triangulation, demonstrates no reverse use-dependency, and has very diminutive probability for inducing TdP (209). In addition to the increased risk of inducing EADs by inappropriate inhibition of repolarization K⫹ channels leading to triangulation, the large muscle mass in ventricles, compared with atria, further adds to the increased propensity for pro-arrhythmic effects of K⫹ channel inhibition. Electrical heterogeneities among cardiac cells can exist along several axes including the transmural, left-right, and apicobasal axes (50). As described earlier, such electrical heterogeneities can constitute a substrate for arrhythmias as prominent as those obtained by anatomical remodeling or infarcts. This type of heterogeneity can also arise from acute myocardial ischemia and chronic myocardial infarction (25, 87, 447). In the literature, transmural voltage gradients in particular have received continued interest. In different species including humans, it has been suggested that the ventricular wall is sufficient in thickness to encompass three different areas of cells named endocardial, mid-myocardial, and epicardial areas (56, 293, 427). Particularly the mid-myocardial cells have been speculated as having prolonged action potentials that can cause electrical heterogeneity during repolarization. This heterogeneity is also recognized as action potential dispersion and can be increased under conditions such as ischemia, bradycardia, and drug application (241, 427). To recapitulate, the research in new pharmacological treatments of ventricular tachycardia and VF has been rather unsuccessful, although it has led to a much better understanding of how to avoid pro-arrhythmic effects of new drugs. The current first-line treatment for ventricular arrhythmia is therefore often devices and classical anti-arrhythmic drugs as adjuncts (83, 84). A detailed description of new attempts is given in the section on pharmacological tools. D. Remodeling One of the consequences of sustained fibrillation or congestive heart failure is that these conditions predispose to further fibrillation by inducing changes in the physiological Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org 631 Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 AF is by far the most common sustained cardiac arrhythmia and accounts for approximately one-third of hospitalizations related to heart rhythm disturbances (129). The overall lifetime risk for getting AF is ⬃25% in the general population (254). In a historical perspective, AF was first described in 1906, and characterized as rapid uncoordinated electrical activation of the atria (130). Several classification systems are now used to describe various forms of AF. These include categories such as acute, paroxysmal, intermittent, constant, chronic, persistent, and permanent. When a patient encounters two or more episodes, the AF is considered recurrent. Should the recurrent AF terminate and revert to sinus rhythm spontaneously within a week, the designation paroxysmal is used. If AF continues for longer than 7 days, it is categorized as persistent. Finally, AF can become permanent. This is a clinical definition for ongoing AF resistant to cardioversion or for very long-term AF for which cardioversion has not been attempted. SCHMITT, GRUNNET, AND OLESEN and anatomical properties of the cardiac tissue. It should be accentuated that fibrillatory events per se can change the overall distribution and function of ion channels in a quantitative and qualitative fashion without evident structural changes. This process is known as electrical remodeling. Hence, we will subsequently give an account of essential changes in ion channel function and distribution as a consequence of remodeling. 1. Atrial remodeling AF is a condition that tends to develop or progress over time. A consequence of especially sustained AF is remodeling of the atria. The changes encompass both structural and electrical alternations. Structural remodeling primarily involves atrial dilatation, hypertrophy, and increased amount and changed distribution of fibrotic tissue not able to conduct electrical signals (60, 61, 71). Electrical remodeling is the consequence of an overall qualitative and quantitative change in ion channel distribution. The combined effects of structural and electrical remodeling result in AF being a self-perpetuating disease if left untreated. The reason for this is that remodeling increases the propensity for triggering events that can initiate future incidences of AF. This will happen in combination with the tissue becoming more vulnerable to electrical disturbances and the development of larger areas of nonconducting tissue that can serve as a substrate for reentry-based AF. Consequently, patients with episodes of paroxysmal AF can progress into persistent and permanent AF, a phenomenon that has been characterized as “AF begets AF” (7, 105, 303, 456). In addition to being a direct outcome of tachypacing and AF, remodeling of atria can also arise as a secondary effect from cardiac diseases that are not directly related to arrhythmia. These could be congestive heart failure and acute myocardial infarction (300). The following description of remodeling focuses on effects on ion channels as a direct consequence of AF. A brief overview of the effects of remodeling is given in FIGURE 10. At first sight remodeling might be perceived as a condition resulting in nothing but an unfavorable outcome. Despite the damaging long-term consequences, however, there are acute beneficial effects associated with remodeling. In AF tissue, atrial contraction is significantly increased due to the high frequent initiation of action potentials and corresponding influx of Ca2⫹. A very consistent observation in 632 Upregulation of K⫹ channels is also a prominent contributor to the action potential shortening associated with tachypacing and AF. Several studies showed increased mRNA and protein levels for Kir2.1 channels that are the main component of IK1 (132, 172, 331, 492). In contrast, IK1 is downregulated or unchanged when remodeling is based on myocardial infarction or congestive heart failure (240, 345). This notion emphasizes the complexity of changes and the importance of the condition that provokes the remodeling. It seems justifiable to hypothesize that there may be different types of AF based on the biological mechanism causing the disease. The upregulation of IK1 during AF will contribute to a further reduction in action potential duration and ERP, which is proarrhythmic. Furthermore, it has been demonstrated that increased IK1 can increase the curvature of spiral waves and thereby stabilize rotors and reentry arrhythmias (74, 373). Also Kir3.1/3.4 responsible for IKACh is affected by remodeling. The constitutive component of this channel is normally tiny, and a significant IKACh normally requires an increased parasympathetic or vagal tone. Vagal activation is proarrhythmic as the IKACh activation leads to a shortening of ERP and stabilization of rotors (81, 220, 480). A reduction of IKACh has been reported in conjunction with AF, but it has also been suggested that the constitute component of IKACh is increased as a consequence of AF remodeling leaving a sustained vagally independent current activity that would be proarrhythmic (104, 438). Remodeling of KV1.5 channels and thereby IKur in the context of AF is more equivocal. There have been reports of both a reduction in current density and of no changes (49, 52, 439, 462). It is a paradox that although only modest or Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 Changes in electrophysiological parameters are due to redistribution and up- or downregulation of various ion channels. The exact outcome of remodeling depends on the underlying disease and differs according to whether it is introduced by atrial tachypacing or by congestive heart failure (300). Remodeling can also differ between atria and ventricles, even when originating from the same condition. The following section is therefore divided into separate paragraphs covering atrial and ventricular remodeling, respectively. different species challenged with AF or tachypacing is a reduction in voltage-gated L-type Ca2⫹ channels, while Ttype channels seem to be unaffected (485). Ca2⫹ overload is potentially lethal, and the downregulation of L-type channels can be seen as a logical response serving to reduce Ca2⫹ influx to the myocytes (48, 49, 77, 462, 472). The acute benefits of decreasing Ca2⫹ overload via a reduction in L-type Ca2⫹ channels is, however, jeopardized in the longer perspective. Reduced Ca2⫹ current will decrease the action potential duration by reducing the plateau phase, meaning a shorter and also more triangulated action potential. This will in theory be proarrhythmic, since ERP is reduced and the risk for EAD-based triggering events is increased. When it comes to an increased propensity for atrial EADs, the situation is not quite as simple. Although triangulation enlarges the vulnerable window for reactivation of L-type Ca2⫹ channels and thereby EADs, the concomitant reduction in channel number will counteract this tendency. Predicting whether the outcome is an increased or decreased risk for EAD-initiated arrhythmia can be difficult, but EADrelated AF is well-described (58, 378). CARDIAC POTASSIUM CHANNELS AND ARRHYTHMIA A Normal Decreased number; increased lateralization, heterogeneity of connexins Remodeled CV INa Substrate for multiple circuit reentry Normal Remodeled APD IKACh,c WL: Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 RP Initiation of reentry ICaL Increased triggering (?) IK1 Ca2+ handling abnormalities Pathophysiology of AF promotion by atrial-tachycardia remodeling B IK1 ATRIAL TACHYCARDIA ICa IKur IKAch AP SHORTENING TRIANGULATION Ito FIGURE 10. Remodeling of ionic currents in atrial fibrillation. A: decreased conduction velocity (CV), decreased effective refractory period (ERP), or a combination of both results in a decrease of the wavelength. CV can be reduced as a consequence of diminished gap junction or subcellular relocation of gap junctions resulting in increased lateralization that will disturb and reduce normal conduction. Changes in the expression patterns of a number of ion channels result in action potential shortening and increased triangulation. For details, see text. [From Nattel et al. (300).] B: ion channel changes associated with atrial remodeling. no changes in IKur have been observed in association with AF, the contribution from this current to repolarization appears very different in AF patients compared with control individuals in sinus rhythm. IKur inhibition in AF patients prolongs action potential duration, while a reduction in action potential duration is observed in sinus rhythm, probably reflecting a changed relative significance of IKur due to up- and downregulation of other channels (78, 90, 359). This truly demonstrates the complexity of remodeling and the importance of considering the entire composition of ion channels when trying to predict the impact of remodeling of a single type of current. In the context of lone AF, it should also be mentioned that some reports have associated loss- of-function mutations in KCNA5 encoding KV1.5 and thereby IKur to increased risk for AF (322, 476), and recently KCNA5 gain-of-function mutations (79) have been reported in a number of lone AF patients. Very recently, Ling and co-workers (70) reported that atrial miRNA-499, which is significantly upregulated in AF, leads to KCa2.3 downregulation by binding of miR-499 to the 3’-UTR of KCNN3, and hence is possibly involved in electrical remodeling in AF. Downregulation of Ito seems to be a consistent finding in relation to remodeled atria (48, 439, 462). A decreased Ito Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org 633 SCHMITT, GRUNNET, AND OLESEN can affect action potential duration both directly and indirectly. As expected from a reduction in repolarizing capacity, action potential duration is increased when Ito is downregulated. This has been demonstrated in in silico studies and in studies employing cardiac atrial myocytes from rabbit and human (275, 463). The indirect mechanism relies on the fast activation that can oppose depolarizing inward Na⫹ currents. In a situation of reduced Ito, the balance between depolarizing and repolarizing currents can change, thereby resulting in increased action potential amplitude (299). Remodeling of the gap junction proteins connexins 40 and 43 is reported in conjunction with tachypacing and AF. There are some discrepancies in the reports concerning the overall expression level of connexins, with data indicating either reduced, enhanced, or unchanged connexin levels (300). From the perspective of affecting AF, the subcellular distribution of connexins might be of more importance than the total expression level, since this could affect conduction velocity to a larger degree. One of the reported effects of AF-induced remodeling is translocation of connexins from intercalated disks to lateral membranes (224, 351). Such subcellular distributional changes will augment the heterogeneity of impulse propagation and thereby probably increase the propensity for AF. Finally, some consequences of remodeling that are not directly related to ion channels deserve to be briefly mentioned. Conditions such as acute ischemia, atrial dilation, and inflammation are likely to contribute to an increased substrate that will prone the maintenance of AF (299). Atrial dilation, especially in the left atria, causes tissue enlargements. As a result, more reentry circuits can be encompassed, which is proarrhythmic per se (190). This condition is observed after tachypacing as well as in heart failure (390). Another important contributor to increased substrates is generation of fibrotic tissue, which is normally encountered in heart failure and is associated with the development of AF (239, 391). 2. Ventricular remodeling A prerequisite for understanding the significance of changes in ion channel activity is to view them in the broader context of native cells, intact muscles, and intact organs. This is exemplified when analyzing ventricular remodeling as a consequence of congestive heart failure. The condition is 634 For K⫹ channels, the most consistent change is a reduction in the phase 1 repolarizing Ito (300). The change is due to reduction in the principal ␣-subunit KV4.3, while the expression of the associated -subunit KChIP2 is unaltered (4, 368, 399, 501). Another denominator of heart failure remodeling seems to be a reduction in the phase 3 repolarizing IK1 encoded by Kir2.1, although findings are not as consistent as observed for Ito (300, 369, 399, 423). Of the two remaining phase 3 repolarization currents, there may be a reduction in IKs, whereas IKr is reported to be unaffected (300, 399). Remodeling of heart failure ventricles causes a few additional changes: reduction in peak INa but increase in persistent INa,late, decreased connexin 43 expression, and decreased pacemaking If current (3, 375, 428, 430, 431, 500). The ventricular reduction of HCN2 and HCN4 subunits conducting If is another example of opposite consequences of remodeling in atria and ventricles, since these subunits are upregulated in failing atria (500). The increased strength of contraction in early remodeling comes at a price of increased risk of arrhythmias for several reasons. The downregulation of the repolarizing reserve K⫹ currents will postpone the phase 3 repolarization phase and increase the vulnerable window for reactivation of voltagegated Ca2⫹ channels. Together with the increased open probability of these Ca2⫹ channels, the overall consequence Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 Nav1.5 channels may also be influenced by remodeling, but few and conflicting results are reported. Reductions have been demonstrated in tachypaced dogs, and a significant 16% decrease in peak INa density has also been seen in atrial right appendage cardiomyocytes isolated from AF patients compared with controls (135, 401). However, other studies on atrial tissue from human AF patients did not observe changes in INa (49, 132). characterized by severe cardiac impairment and the inability to meet the requirements for proper circulation (300). Heart failure significantly increases the risk of ventricular arrhythmias (218). Approximately half of the death incidents in heart failure patients are related to ventricular arrhythmias, and on top of this atrial arrhythmias are a common companion to heart failure (112). Whereas the hallmark of AF was shortened and triangulated atrial action potentials, the characteristic feature of ventricular remodeling in heart failure is a prolonged ventricular action potential. In the early phase, this will help to partly compensate for reduced ventricular function by improving the contraction strength (370). The prolongation is primarily due to changes in voltage-gated Ca2⫹ channels and K⫹ channels. Overall, the conductance through voltage-gated Ca2⫹ channels is increased. The number of channels is actually reduced, but this is more than compensated for by an increased open probability (73). It should also be mentioned that changes in Ca2⫹ handling proteins other than ion channels occur in heart failure. Expression of the Na⫹/Ca2⫹ exchanger NXC is enhanced by heart failure, while the expression and function of the Ca2⫹-ATPase SERCA2a located in sarcoplasmic reticulum is downregulated (374, 383, 399). Finally, there is an increased Ca2⫹ leak from the sarcoplasmic reticulum to the cytosol due to hyperphosphorylation of Ca2⫹-release channels or RyRs (388, 479). CARDIAC POTASSIUM CHANNELS AND ARRHYTHMIA is a marked increase risk for EADs that can trigger arrhythmic events (193, 242, 314). As IKr is the main repolarizing current not downregulated in heart failure myocardium, there is an excessive risk of action potential prolongation, triangulation, and arrhythmia when exposing patients to drugs that inhibit this channel. IKr inhibition is an unintended property of many drugs used in the clinic, and extra care should be taken with such compounds in the event of heart failure (424). From the perspective of prolonged ventricular action potentials and increased risk of EADs, heart failure can be viewed as a kind of acquired LQTS (76). Furthermore, the remodeling of Ca2⫹ handling proteins other than ion channels contributes to the proarrhythmic effects. The combination of increased Ca2⫹ leak from sarcoplasmic reticulum (SR), reduced SR uptake due to decrease SERCA2a function, and increased Na⫹/Ca2⫹ exchanger (NCX) function is problematic for two reasons. First, the general reduction in SR Ca2⫹ content will decrease contractile function. Second, the increased cytosolic Ca2⫹ concentration in combination with increased amount of NCX will increase cellular Ca2⫹ efflux at the expense of increased Na⫹ uptake. This electrogenic transport can provoke DADs and trigger arrhythmias (300). Finally, the increase in the persistent Na⫹ current INa,late will participate in the prolongation of ventricular action potentials and increase the risk for EADs (428, 430). The downregulation of HCN subunits and thereby If can cause bradycardia and contribute to cardiac decompensation (6). The use of pharmacological tool compounds has been of immense value for understanding the physiology of cardiac electrical activity and pathophysiology of cardiac arrhythmias. The following section focuses on initiatives targeting K⫹ channels and introduces the ideas behind some of the novel approaches for developing antiarrhythmic drugs and the experiences with them. The main body will be on strategies for targeting AF, but an outline on new pharmacological treatment of VF by K⫹ channel modulation is also included. A. Strategies for Treatment of AF The two principal strategies for treating AF are rhythm and rate control, respectively. The aim of rhythm control is to restore and maintain normal sinus rhythm. In contrast, rate control aims to reduce the ventricular impact of AF by protecting the ventricles from the fast atrial electrical activity. In other words, rate control will leave the atria fibrillating and prevent excessive conduction from atria to ventricles. This is accomplished by the application of beta blockers, calcium channel blockers, digoxin, or amiodarone that prolongs refractoriness in the AV node and slows conduction through the AV node. Considering these aims, it would be expected that rhythm control, which targets AF rather than its downstream consequences, would be preferential to rate control. This has, however, not been justified by trials such as AFFIRM, RACE, PIAF, STAF, and HOT CAFÉ that compared the ability of rhythm versus rate control to reduce hard end points such as stroke and death (68, 137, 181, 324, 466). The nonsuperiority of rhythm control in these trials is surprising, since it could be expected that reversion of AF to normal sinus rhythm would decrease blood stasis, reduce the risk of stroke, and concomitantly improve hemodynamic parameters. An explanation for the nonsuperiority of rhythm versus rate control might be that the RACE and AFFIRM trials did not include younger symptomatic AF patients without underlying heart diseases. Another reason might be the applied strategy for analyzing the data. Based on the AFFIRM information, it was demonstrated for example that sinus rhythm was an independent predictor of reducing the risk of death, but simultaneously increased overall mortality (hazard ratio ⫽ 1.49) (86, 337). The explanation for this overall increase in mortality is likely due to the ventricular proarrhythmic effects associated with some of the drugs developed for rhythm control, as demonstrated by the CAST and SWORD study (110, 119, 440). In a recent study on 26,130 Canadian AF patients with a longer follow-up, the relative advantages of the two treatment paradigms were time dependent. The rhythm control group Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org 635 Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 The reduction of IK1 has also been associated with proarrhythmia due to increased risk of DADs due to the important function of IK1 in setting the resting membrane potential in the diastole. Under normal conditions, the high open probability of IK1 at resting membrane potential results in a correspondingly low membrane resistance. The low resistance will decrease the amplitude of abnormal depolarizations such as DADs. In contrast, when IK1 is reduced, the diastolic membrane resistance is increased, and a DAD will result in larger current amplitude with higher chance of reaching the threshold for evoking a premature action potential (350). These physiological mechanisms emphasize the somewhat ignored fact that a high diastolic K⫹ conductance can be a very important antiarrhythmic parameter, both to decrease the likelihood for DADs to elicit inappropriate action potentials, as well as to set the stage for the next systole by affecting the release from inactivation of Na⫹ channels. The antiarrhythmic effect of some of the recent activators of KV11.1 channels and thereby IKr might be explained by increased diastolic K⫹ conductance (169 – 171). Importantly, this could also be a parameter that discriminates application of KV11.1 activators from short QT mutations such as KV11.1-N588K (154). IV. PHARMACOLOGICAL TOOLS FOR ADDRESSING CARDIAC Kⴙ CHANNEL FUNCTION SCHMITT, GRUNNET, AND OLESEN showed a slight increase in mortality during the first 6 months, equal mortality until year 4, and a lower mortality than the rate control group after 5 years, indicating a long-term superiority of rhythm control (189). For these reasons, the search for atrial-selective drugs without extracardiac toxicity and ventricular proarrhythmic effects is the favorable approach when designing new antiarrhythmic drugs. The current guidelines also still support restoration of sinus rhythm in younger AF patients as first-line treatment (128, 129). Substantial preclinical and clinical information has been gathered for dronedarone, one of the anti-arrhythmic drug newcomers. While this drug was developed to preserve the efficacy of amiodarone and reduce adverse effects by deleting iodine in the molecule, the outcome remained equivocal. Compared with amiodarone, dronedarone seems less efficacious in a clinical setting. The clinical study ATHENA comprised patients with paroxysmal or persistent AF (179). Compared with the placebo group, dronedarone resulted in significant reduction in cardiovascular admissions and mortality (180). The DIONYSOS study included an end point of direct comparison of amiodarone and dronedarone. Dronedarone turned out to be less efficacious in patients with persistent AF, but amiodarone had the advantage of better tolerance (177). Noteworthy, the United States Food and Drug Administration (FDA) recently issued a safety announcement stating that the use of dronedarone can be associated with hepatic failure. Furthermore, dronedarone is counter-indicated for heart failure patients due to increased premature death in this patient population, as demonstrated in the preterminated ANDROMEDA study (222). The diverse experiences with the development of dronedarone emphasize the need for careful selection of new antiarrhythmic drugs and thorough preclinical understanding of mode of action. Better patient stratification might also help new antiarrhythmic drugs to succeed in passing clinical development. Current drug discovery and development activities for the treatment of AF target atrial-specific K⫹ channels or aim at selective inhibition of atrial Na⫹ channels. This latter ap- 636 1. IKur blockers Functional IKur is generally believed to be absent from ventricles (13). Thus blocking IKur has been perceived as an attractive strategy for developing a new drug against AF (125). However, inhibiting IKur by low concentrations of 4-aminopyrimidine or AVE0118 in atrial trabeculae from individuals in sinus rhythm led to shortened action potentials. This counterintuitive finding is explained by an elevated plateau potential that allows for more prominent IKr and IKs activation and thereby accelerated repolarization (454). The results emphasize the importance of addressing modulations of ion channels in the perspective of the orchestrated function of all other ion channels in play. This is further underscored by the fact that inhibition of IKur causes different effects in remodeled tissue from AF patients. Here the triangulated action potential gives room for less elevated plateau potentials, and consequently IKur inhibition increases action potential duration (454). It is still an open question whether a pure IKur blocker will be sufficient for AF suppression. It is worth keeping in mind that chronic AF in humans might reduce KV1.5 expression and thereby IKur density (78, 439). Also it should be mentioned that the importance of IKur in humans seems to be less pronounced than suggested from preclinical animal models (336). In addition, IKur density is diminished at high atrial heart rates as seen in AF (122). Furthermore, a safety concern is evident since it has been demonstrated that loss-of-function mutation in KCNA5 giving rise to reduced IKur is associated with familial AF (322, 476). A number of KV1.5 channel inhibitors have been or are in development for treating AF, and the applicability of IKur inhibition as a valid approach for treating AF is likely to be clarified in the future. Some of these compounds are AVE0118 (IC50 1.1 M), which also has affinity for Ito and IKAch (142); DPO-1 as a use-dependent open IKur channel blocker with some affinity for IK1 and IKs (230, 361); and more selective compounds such as XEN-D0101 and XEN-D0103 with human atrial IKur IC50 values of 241 and 154 nM, respectively (125). It has generally proven difficult to obtain good selectivity among Kv channel subtypes, as many drugs interact with highly conserved residues in the central cavity. Using alanine-scanning on KV1.x channels, new drug binding sites have been identified on pockets formed by the backsides of several transmembrane segments, indicating a possible avenue for overcoming this problem (277). Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 Triggers and reentry propagation of electrical signals in a changed substrate are the key mechanisms of AF as mentioned above. Inhibition of K⫹ channels has proven effective in prolongation of the effective refractory periods and interruption of reentry-based arrhythmias. Such compounds constitute the class III antiarrhythmics according to the Vaughan-Williams classification. The typical class III drugs are dofetilide and ibutilide, but sotalol and multichannel inhibitors such as amiodarone and dronedarone also exhibit class III properties (69, 235, 344). All these drugs inherently influence both atrial and ventricular signaling, and it is evident that many of their adverse effects would be reduced if they solely targeted atrial ion channels. proach takes advantage of the fact that resting membrane potential is more positive in atria compared with ventricles. Consequently, Na⫹ channels are more inactivated in atria, which can be exploited to generate state-dependent and thereby selective inhibitors of atrial Na⫹ channels. Since this review focuses on K⫹ channels, no further introduction to state-dependent Na⫹ channel inhibition will be included. More comprehensive overviews can be found in References 16 and 153. CARDIAC POTASSIUM CHANNELS AND ARRHYTHMIA 2. IKAch blockers 3. Other concepts Preclinical evidence points to Ca2⫹-activated KCa2.x channels as a target that possesses functional atrial selectivity, and KCa2.x channel inhibition has demonstrated to be efficient in terminating acutely induced AF in various animal models (101, 102, 396). These studies have recently been extended in dogs with atrial fibrillation induced by atrial tachypacing. Atrial tachypacing increased whole cell and single channel KCa2.x current. Pharmacological inhibition of KCa2.x increased action potential duration in an atrialselective manner and demonstrated antiarrhythmic properties in dogs (357). For a comprehensive overview or recent development of drugs targeting AF, see also Reference 153. Some attempts have also been made to investigate the antiarrhythmic potential of modulating the IK1 current. However, since IK1 is by no means an atrial-selective target, it is debated whether increased or decreased activity will be beneficial. Various studies demonstrated that increasing IK1 is proarrhythmic due to stabilizing rotors (252, 446). However, antiarrhythmic effects have also been observed after IK1 activation (252). B. Strategies for Treatment of VF In recent years, new developments of K⫹ channel modulators have been sparse as a consequence of the proarrhythmia associated with traditional class III antiarrhythmics and the current first-line treatment with implantable electronic devices (84, 86, 110, 119). However, there have been some preclinical approaches investigating the idea of an increased repolarization reserve as a new principle for treating ventricular arrhythmias. Test molecules have been developed activating all phase 3 repolarizing K⫹ currents IKr, IKs, and IK1 carried by KV11.1, KV7.1, and Kir2.1, respectively, but it is primarily activators of KV11.1 that have been thoroughly investigated. KV11.1 channels show very characteristic inactivation properties with fast inactivation upon depolarization and fast recovery from inactivation upon repolarization (FIGURE 11A). Many KV11.1 channel activators affect these properties by reducing inactivation or promoting recovery from inactivation. A reduction in relative inactivation, being equal to a rightward shifted inactivation curve, will increase the overall KV11.1 current. Compounds with such properties include NS1643 and NS3623 (both NeuroSearch), PD-307243 (Pfizer), and A-935142 (Abbott) (151, 168, 169, 411, 498). RPR260243 (Sanofi-Aventis) utilizes a different mechanism of action, as it slows KV11.1 channel closure (deactivation) corresponding to an increase in macroscopic IKr (203). Importantly, the pharmacological effect of these compounds is different from the biophysical properties revealed for a KV11.1 channel mutation that has been associated with the SQTS (154). The antiarrhythmic properties of this new compound class were demonstrated experimentally ex vivo and in vivo. In patch-clamp studies, NS1643 and NS3623 shorten ventricular action potentials when recorded from acutely isolated single cells (FIGURE 11B), and the same notion holds true in isolated papillary muscle. Importantly, an increased ability to withstand triggering activity in the form of mimicked EADs was also demonstrated (100, 168, 169). Similar results have been obtained with A-935142 in guinea pigs and dogs (411). PD-118057 has also been tested ex vivo in iso- Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org 637 Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 The K⫹ current IKAch composed of Kir3.1/Kir3.4 subunits is another atrial-selective target that has not been as extensively investigated as KV1.5 channels, but evidence has been accumulating, particularly in recent years, for this to be an interesting target (358). As for KV1.5, the rationale for inhibiting this channel is to obtain selective prolongation of the atrial effective refractory periods. Interesting IKAch inhibitors are NTC-801 with an IC50 of 0.7 nM as well as NIP-151 and NIP-142 with IC50 values of 1.6 and 640 nM, respectively (269, 297, 416, 418). Noteworthy, many multichannel inhibitors developed for treatment of AF display affinity for IKAch. These include amiodarone, dronedarone, AVE0118, E-4031, vernakalant, sotalol, and flecainide (121, 142, 163, 288, 448). The idea about an increased repolarization reserve as an antiarrhythmic strategy appeared as a logical extension of the proarrhythmic effects observed by prolonging the ventricular action potential with class III drugs. It was demonstrated that proarrhythmia is primarily due to the inhibition of KV11.1 channels, and it became a prerequisite in drug development to screen potential drug candidates for their ability to inhibit this channel. A number of high-throughput assays were developed for the purpose, and as a result of screening hundreds of thousands of compounds for KV11.1 inhibition, a number of activators of this channel also appeared. Several companies pursued the idea that the conditions or diseases associated with prolonged QT intervals and underlying ventricular action potential prolongations could be normalized by the application of KV11.1 channel activators. From a theoretical perspective, the primary antiarrhythmic effects of such compounds would be shortening of the action potential duration and reduced triangulation leading to narrowing of the vulnerable window leaving less room for EADs that can trigger an arrhythmia. Since KV11.1 channels deactivate slowly during the early phase 4, an increased current will further help antagonize DADs here. Decrease in electronic heterogeneity could be another beneficial parameter. Relevant indications could be congenital and acquired long QT syndromes, heart failure, and prolonged QT in association with AF, ischemia, diabetes, etc. SCHMITT, GRUNNET, AND OLESEN A K+ Slow Fast K+ Slow Fast K+ Closed Open Inactivated B b Control 50 Voltage (mV) 50 0 –50 0 –50 –100 –100 0.0 0.4 c 0.8 Time (s) 1.2 0.0 0.4 d NS1643 100 50 50 0 0.8 Time (s) 1.2 NS1643 100 Voltage (mV) Voltage (mV) Control 100 0 –50 –50 –100 –100 0.0 0.4 0.8 Time (s) 1.2 0.0 0.4 0.8 Time (s) 1.2 0.0 0.4 0.8 Time (s) 1.2 FIGURE 11. KV11.1 pharmacology. A: KV11.1 gating kinetics. B: activation of KV11.1 with the tool compound NS1643 in acutely isolated cardiomyocytes from guinea pig. The response of cardiomyocytes to simulated EADs was assessed after an elicited action potential; a train of 10 –20 50% subthreshold stimuli was applied either at control situations after full repolarization (a and c) or to induce EADs at APD90 (b and d). In the absence of NS1643, subthreshold stimuli applied at APD90 were able to induce EAD events every time the protocol was applied, and prolongation of the action potential was continued even after cessation of current injection (b). Upon application of 10 M NS1643, the premature train of stimuli was not able to trigger any abnormal activity, and application of the drug thus led to a stable action potential (d). Superimposed traces for subthreshold stimuli applied at APD90 in the absence or presence of NS1643 are shown in e, left. Current injection protocol is shown in e, right. [B from Hansen et al. (168).] e Voltage (mV) 100 50 0 –50 –100 638 Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 Voltage (mV) a 100 CARDIAC POTASSIUM CHANNELS AND ARRHYTHMIA lated rabbit and canine wedge preparations with somewhat opposing results. A clear antiarrhythmic effect was observed in the rabbit wedge and included the expected action potential shortening, but also reduced dispersion. In contrast, canine wedge studies resulted in increased dispersion and pacing-induced polymorphic ventricular tachycardia (334, 498). In intact isolated hearts, NS3623 has demonstrated antiarrhythmic properties such as a decrease in QT variability, and a tendency towards reduced global dispersion when addressed as a reduction in Tpeak-Tend intervals, as well as a marked reduction in number of extrasystoli (170). For KV7.1 channels and IKs, only limited experience is available on pro- or antiarrhythmic effects of increasing this current, with the benzodiazepine R-L3 being the central compound. R-L3 reverses pharmacologically induced type 2 LQTS (induced by IKr inhibition), shortens action potential duration, and decreases triangulation. The compound activates heterologously expressed KV7.1 channels, but has no effect on the KV7.1/KCNE1 complex that mimics native IKs (371, 384). R-L3 also reduces the effective refractory period, which might indicate proarrhythmic activity by increasing the risk for atrial fibrillation (310). More comprehensive overviews of exact mode of actions and anti- and proarrhythmic properties of KV11.1 and KV7.1 activators are given in References 155 and 162. Another innovative approach to address cardiac ion channels has been the development of activators of Ito. Applying the tool compound NS5806 (NeuroSearch) in isolated canine ventricular myocytes increased peak currents significantly and slowed inactivation kinetics. Furthermore, in V. CONCLUSIONS AND PERSPECTIVES The molecular studies of the diverse cardiac potassium channel subtypes have allowed a detailed picture of their individual structures and regulatory mechanisms to emerge. By addressing the significance of accessory subunits and influence of posttranslational modifications, it has also been widely possible to translate these molecular correlates into the endogenous cardiac K⫹ currents. Yet most of the stateof-the-art techniques employ heterologously expressed channels or depict snap shots of cardiac tissue, and hence only partially reflect processes in vivo. Accordingly, a larger challenge that still remains is to understand how these “actors in the still picture” behave in dynamic play and change their function relative to each other both in the short term (changes in heart rate, sympathetic tone, Ca2⫹ level, posttranslational modifications) and long term (epigenetic changes, remodeling). These changes of the basic biological conditions of the tissue determine whether the stable heart rhythm is maintained or an arrhythmia is triggered. We have in this review addressed the concerted action of the repolarizing cardiac K⫹ currents, while also seeing them as individual currents. The use of selective pharmacological tools, both channel-subtype-specific toxins and organic molecules, has proven valuable to dissect the role of the individual channels in intact organisms. One result of these pharmacological studies combined with expression analysis and other functional investigations is the identification and Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org 639 Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 In vivo NS3623 has been tested in anesthetized and conscious guinea pigs. In line with theoretical considerations, the measured and corrected QT interval was reduced in both conditions. Furthermore, NS3623 reverses drug-induced QT prolongation (171). In rabbits, NS1643 has demonstrated antiarrhythmic properties in a pharmacological and nonpharmacological model for ventricular fibrillation. In the pharmacological model, the combined proarrhythmic effect of the class III drug dofetilide together with the ␣1-adrenergic receptor agonist methoxamine could be prevented by NS1643. In a more physiological model, atrioventricular block and subsequent 3-wk bradypacing resulted in ventricular extrasystoli and short runs of TdP like arrhythmias. Both conditions could be prevented by NS1643 (103). Although the models are promising, it should also be mentioned that proarrhythmic effects have been associated with KV11.1 channel activators, although some of these might be unspecific effects (162, 259). In terms of therapeutic application, KV11.1 channel activators probably have the highest chance of success in conditions with extensive action prolongation that can subsequently be normalized by this new compound class. coronary-perfused wedge preparations, NS5806 recapitulated the features of Brugada Syndrome phenotype visible as increase in phase 1 and notch amplitudes (62). The effect was later shown to be dependent on the presence of the Ito subunit K channel interacting protein (KChIP) 2 (65, 263). Accordingly, the transmural KChIP2 gradient across the ventricular wall led to differential effects of NS5806 in epi-, mid-, and endocardial cells (65). A study comparing NS5806 in canine atrial and ventricular myocytes showed a different response in the atria, where the addition of drug had only mild effects on phase 1. Instead, the upstroke velocity was significantly decreased, which could be ascribed to a strong inhibition of INa in the atria (63). Although NS5806 in addition to NaV1.5 does affect other potassium channels such as KV1.4 and KV1.5, it represents an interesting tool compound in the context of heart failure (HF). HF, amongst others, is characterized by an action potential prolongation due to remodeling the repolarizing currents Ito and IKs (reviewed in Ref. 88). The reduction in Ito is due to the reduction of KV4.3 channels (202). NS5806 was used in a tachypaced-induced canine model of heart failure and was able to restore phase 1 action potential morphology in wedge preparations from HF dogs (85). Despite the risk of being proarrhythmic, this tool compound provided a proof-of-concept that Ito activators might be useful for treatment of HF. SCHMITT, GRUNNET, AND OLESEN verification of the fairly atrial-specific repolarizing currents IKur, IKAch, IKCa, and IK2P. The role of the first two channels in the atria has been known for a number of years, whereas the significance of the latter two is new and may pave the way for new treatment paradigms for AF. GRANTS Over the past 20 years, the mutations in single cardiac ion channels have been a rich source of information about the role of each channel subtype in arrhythmogenesis and have fostered the concept of monogenetic arrhythmic diseases. The significance of these variants is currently being rewritten on the background of the exome data becoming available from large cohorts showing their presence in the general population and calling for a stricter interpretation. DISCLOSURES The best translational studies to provide us with an understanding of the specific role of a given K⫹ channel on the background of the patient’s own biological background use fresh human cardiac tissue, often removed in open chest operations with extracorporal circulation. To investigate specific genetic variants, the use of induced pluripotent stem cells generated from the patient’s skin biopsies or blood has fostered hope for the development of true personalized medicine. ACKNOWLEDGMENTS We thank Jonas Bille Nielsen and Lasse Skibsbye (both from the Danish National Foundation Centre for Cardiac Arrhythmia) for kindly providing ECG recordings and human cardiac action potentials, respectively. N. Schmitt and M. Grunnet contributed equally to the present work. Address for reprint requests and other correspondence: S.-P. Olesen, The Danish National Research Foundation Centre for Cardiac Arrhythmia, Dept. of Biomedical Sciences, Faculty of Health and Medical Sciences, Univ. of Copenhagen, Copenhagen, Denmark (e-mail: [email protected]. dk). 640 M. Grunnet is employed at Lundbeck A/S (Valby, Denmark) and Acesion Pharma (Copenhagen, Denmark). REFERENCES 1. Aarnoudse AJLHJ, Newton-Cheh C, de Bakker PIW, Straus SMJM, Kors JA, Hofman A, Uitterlinden AG, Witteman JCM, Stricker BHC. Common NOS1AP variants are associated with a prolonged QTc interval in the Rotterdam Study. Circulation 116: 10 –16, 2007. 2. Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, Keating MT, Goldstein SA. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 97: 175–187, 1999. 3. Akar FG, Spragg DD, Tunin RS, Kass DA, Tomaselli GF. Mechanisms underlying conduction slowing and arrhythmogenesis in nonischemic dilated cardiomyopathy. Circ Res 95: 717–725, 2004. 4. Akar FG, Wu RC, Juang GJ, Tian Y, Burysek M, Disilvestre D, Xiong W, Armoundas AA, Tomaselli GF. Molecular mechanisms underlying K⫹ current downregulation in canine tachycardia-induced heart failure. Am J Physiol Heart Circ Physiol 288: H2887– H2896, 2005. 5. Albesa M, Grilo LS, Gavillet B, Abriel H. Nedd4-2-dependent ubiquitylation and regulation of the cardiac potassium channel hERG1. J Mol Cell Cardiol 51: 90 –98, 2011. 6. Alboni P, Brignole M, Menozzi C, Scarfò S. Is sinus bradycardia a factor facilitating overt heart failure? Eur Heart J 20: 252–255, 1999. 7. Allessie M, Ausma J, Schotten U. Electrical, contractile and structural remodeling during atrial fibrillation. Cardiovasc Res 54: 230 –246, 2002. 8. Allessie MA, Bonke FI, Schopman FJ. Circus movement in rabbit atrial muscle as a mechanism of trachycardia. Circ Res 33: 54 – 62, 1973. 9. Allessie MA, Bonke FI, Schopman FJ. The mechanism of supraventricular tachycardia induced by a single premature beat in the isolated left atrium of the rabbit. I. Circus movement as a consequence of unidirectional block of the premature impulse. Recent Adv Stud Card Struct Metab 5: 303–308, 1975. 10. Allessie MA, Bonke FI, Schopman FJ. Circus movement in rabbit atrial muscle as a mechanism of tachycardia. II. The role of nonuniform recovery of excitability in the occurrence of unidirectional block, as studied with multiple microelectrodes. Circ Res 39: 168 –177, 1976. 11. Allessie MA, Bonke FI, Schopman FJ. Circus movement in rabbit atrial muscle as a mechanism of tachycardia. III. The “leading circle” concept: a new model of circus movement in cardiac tissue without the involvement of an anatomical obstacle. Circ Res 41: 9 –18, 1977. 12. Amin AS, Giudicessi JR, Tijsen AJ, Spanjaart AM, Reckman YJ, Klemens CA, Tanck MW, Kapplinger JD, Hofman N, Sinner MF, Müller M, Wijnen WJ, Tan HL, Bezzina CR, Creemers EE, Wilde AAM, Ackerman MJ, Pinto YM. Variants in the 3’ untranslated region of the KCNQ1-encoded Kv7.1 potassium channel modify disease severity in patients with type 1 long QT syndrome in an allele-specific manner. Eur Heart J 33: 714 –723, 2012. 13. Amos GJ, Wettwer E, Metzger F, Li Q, Himmel HM, Ravens U. Differences between outward currents of human atrial and subepicardial ventricular myocytes. J Physiol 491: 31–50, 1996. 14. Andreasen C, Refsgaard L, Nielsen JB, Sajadieh A, Winkel BG, Tfelt-Hansen J, Haunsø S, Holst AG, Svendsen JH, Olesen MS. Mutations in genes encoding cardiac ion channels previously associated with sudden infant death syndrome (SIDS) are present with high frequency in new exome data. Can J Cardiol 29: 1104 –1109, 2013. Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 The limited penetrance of some of the disease-causing mutations and the fact that both gain-of-function and loss-offunction mutations in the same channel can cause a similar disease indicate that there are conditions of the cardiac tissue we probably need to take into consideration for each patient to understand the outcome of a changed K⫹ channel function. Great effort is made to generate various disease models in animals to clarify this issue. Yet, because the distinct and complex electrophysiological properties of cardiomyocytes differ among species, the use of isolated cardiomyocytes from model animals has limitations. Since certain smaller animals are quite dissimilar to humans, especially from a cardiac point of view, extending the conclusions to human arrhythmogenesis is applicable only to a limited degree. N. Schmitt and S.-P. Olesen are funded by a framework grant from the Danish National Research Foundation. CARDIAC POTASSIUM CHANNELS AND ARRHYTHMIA 15. Angelo K, Jespersen T, Grunnet M, Nielsen MS, Klaerke DA, Olesen SP. KCNE5 induces time- and voltage-dependent modulation of the KCNQ1 current. Biophys J 83: 1997–2006, 2002. 36. Benjamin EJ, Wolf PA, D’Agostino RB, Silbershatz H, Kannel WB, Levy D. Impact of atrial fibrillation on the risk of death: the Framingham Heart Study. Circulation 98: 946 –952, 1998. 16. Antzelevitch C, Burashnikov A. Atrial-selective sodium channel block as a novel strategy for the management of atrial fibrillation. J Electrocardiol 42: 543–548, 2009. 37. Bennett PB, Yazawa K, Makita N, George AL Jr. Molecular mechanism for an inherited cardiac arrhythmia. Nature 376: 683– 685, 1995. 17. Antzelevitch C. Ionic, molecular, and cellular bases of QT-interval prolongation and torsade de pointes. Europace 9 Suppl 4: iv4 –15, 2007. 38. Benson MD, Li QJ, Kieckhafer K, Dudek D, Whorton MR, Sunahara RK, Iñiguez-Lluhí JA, Martens JR. SUMO modification regulates inactivation of the voltage-gated potassium channel Kv1.5. Proc Natl Acad Sci USA 104: 1805–1810, 2007. 18. Arking DE, Khera A, Xing C, Kao WHL, Post W, Boerwinkle E, Chakravarti A. Multiple independent genetic factors at NOS1AP modulate the QT interval in a multi-ethnic population. PloS One 4: e4333, 2009. 19. Arking DE, Pfeufer A, Post W, Kao WHL, Newton-Cheh C, Ikeda M, West K, Kashuk C, Akyol M, Perz S, Jalilzadeh S, Illig T, Gieger C, Guo CY, Larson MG, Wichmann HE, Marbán E, O’Donnell CJ, Hirschhorn JN, Kääb S, Spooner PM, Meitinger T, Chakravarti A. A common genetic variant in the NOS1 regulator NOS1AP modulates cardiac repolarization. Nat Genet 38: 644 – 651, 2006. 21. Ashmole I, Goodwin PA, Stanfield PR. TASK-5, a novel member of the tandem pore K⫹ channel family. Pflügers Arch Eur J Physiol 442: 828 – 833, 2001. 22. Attali B, Lesage F, Ziliani P, Guillemare E, Honoré E, Waldmann R, Hugnot JP, Mattéi MG, Lazdunski M, Barhanin J. Multiple mRNA isoforms encoding the mouse cardiac Kv1–5 delayed rectifier K⫹ channel. J Biol Chem 268: 24283–24289, 1993. 23. Attwell D, Cohen I, Eisner D, Ohba M, Ojeda C. The steady state TTX-sensitive (“window”) sodium current in cardiac Purkinje fibres. Pflügers Arch Eur J Physiol 379: 137–142, 1979. 24. Baczkó I, Husti Z, Lang V, Leprán I, Light PE. Sarcolemmal KATP channel modulators and cardiac arrhythmias. Curr Med Chem 18: 3640 –3661, 2011. 25. De Bakker JMT, Stein M, van Rijen HVM. Three-dimensional anatomic structure as substrate for ventricular tachycardia/ventricular fibrillation. Heart Rhythm 2: 777–779, 2005. 40. Bers DM, Perez-Reyes E. Ca channels in cardiac myocytes: structure and function in Ca influx and intracellular Ca release. Cardiovasc Res 42: 339 –360, 1999. 41. Bers DM. Cardiac excitation-contraction coupling. Nature 415: 198 –205, 2002. 42. Berthet M, Denjoy I, Donger C, Demay L, Hammoude H, Klug D, Schulze-Bahr E, Richard P, Funke H, Schwartz K, Coumel P, Hainque B, Guicheney P. C-terminal HERG mutations: the role of hypokalemia and a KCNQ1-associated mutation in cardiac event occurrence. Circulation 99: 1464 –1470, 1999. 43. Besana A, Barbuti A, Tateyama MA, Symes AJ, Robinson RB, Feinmark SJ. Activation of protein kinase C epsilon inhibits the two-pore domain K⫹ channel, TASK-1, inducing repolarization abnormalities in cardiac ventricular myocytes. J Biol Chem 279: 33154 –33160, 2004. 44. Bittner S, Budde T, Wiendl H, Meuth SG. From the background to the spotlight: TASK channels in pathological conditions. Brain Pathol Zurich Switz 20: 999 –1009, 2010. 45. Boettger T, Braun T. A new level of complexity: the role of microRNAs in cardiovascular development. Circ Res 110: 1000 –1013, 2012. 46. Bond CT, Maylie J, Adelman JP. Small-conductance calcium-activated potassium channels. Ann NY Acad Sci 868: 370 –378, 1999. 47. Bond CT, Pessia M, Xia XM, Lagrutta A, Kavanaugh MP, Adelman JP. Cloning and expression of a family of inward rectifier potassium channels. Receptors Channels 2: 183–191, 1994. 26. Balse E, Steele DF, Abriel H, Coulombe A, Fedida D, Hatem SN. Dynamic of ion channel expression at the plasma membrane of cardiomyocytes. Physiol Rev 92: 1317– 1358, 2012. 48. Bosch RF, Scherer CR, Rüb N, Wöhrl S, Steinmeyer K, Haase H, Busch AE, Seipel L, Kühlkamp V. Molecular mechanisms of early electrical remodeling: transcriptional downregulation of ion channel subunits reduces I(Ca,L) and I(to) in rapid atrial pacing in rabbits. J Am Coll Cardiol 41: 858 – 869, 2003. 27. Barbuti A, Ishii S, Shimizu T, Robinson RB, Feinmark SJ. Block of the background K⫹ channel TASK-1 contributes to arrhythmogenic effects of platelet-activating factor. Am J Physiol Heart Circ Physiol 282: H2024 –H2030, 2002. 49. Bosch RF, Zeng X, Grammer JB, Popovic K, Mewis C, Kühlkamp V. Ionic mechanisms of electrical remodeling in human atrial fibrillation. Cardiovasc Res 44: 121–131, 1999. 28. Barfod ET, Moore AL, Lidofsky SD. Cloning and functional expression of a liver isoform of the small conductance Ca2⫹-activated K⫹ channel SK3. Am J Physiol Cell Physiol 280: C836 –C842, 2001. 29. Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature 384: 78 – 80, 1996. 30. Bean BP. Two kinds of calcium channels in canine atrial cells. Differences in kinetics, selectivity, and pharmacology. J Gen Physiol 86: 1–30, 1985. 50. Boukens BJD, Christoffels VM, Coronel R, Moorman AFM. Developmental basis for electrophysiological heterogeneity in the ventricular and outflow tract myocardium as a substrate for life-threatening ventricular arrhythmias. Circ Res 104: 19 –31, 2009. 51. Boyle WA, Nerbonne JM. Two functionally distinct 4-aminopyridine-sensitive outward K⫹ currents in rat atrial myocytes. J Gen Physiol 100: 1041–1067, 1992. 52. Brandt MC, Priebe L, Bohle T, Sudkamp M, Beuckelmann DJ. The ultrarapid and the transient outward K⫹ current in human atrial fibrillation. Their possible role in postoperative atrial fibrillation. J Mol Cell Cardiol 32: 1885–1896, 2000. 31. Beaumont J, Davidenko N, Davidenko JM, Jalife J. Spiral waves in two-dimensional models of ventricular muscle: formation of a stationary core. Biophys J 75: 1–14, 1998. 53. Bredt DS, Wang TL, Cohen NA, Guggino WB, Snyder SH. Cloning and expression of two brain-specific inwardly rectifying potassium channels. Proc Natl Acad Sci USA 92: 6753– 6757, 1995. 32. Béguin P, Nagashima K, Nishimura M, Gonoi T, Seino S. PKA-mediated phosphorylation of the human K(ATP) channel: separate roles of Kir6.2 and SUR1 subunit phosphorylation. EMBO J 18: 4722– 4732, 1999. 54. Brohawn SG, Campbell EB, MacKinnon R. Domain-swapped chain connectivity and gated membrane access in a Fab-mediated crystal of the human TRAAK K⫹ channel. Proc Natl Acad Sci USA 110: 2129 –2134, 2013. 33. Behm-Ansmant I, Kashima I, Rehwinkel J, Saulière J, Wittkopp N, Izaurralde E. mRNA quality control: an ancient machinery recognizes and degrades mRNAs with nonsense codons. FEBS Lett 581: 2845–2853, 2007. 55. Brugada R, Hong K, Dumaine R, Cordeiro J, Gaita F, Borggrefe M, Menendez TM, Brugada J, Pollevick GD, Wolpert C, Burashnikov E, Matsuo K, Wu YS, Guerchicoff A, Bianchi F, Giustetto C, Schimpf R, Brugada P, Antzelevitch C. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation 109: 30 –35, 2004. 34. Bellocq C, van Ginneken ACG, Bezzina CR, Alders M, Escande D, Mannens MMAM, Baró I, Wilde AAM. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation 109: 2394 –2397, 2004. 35. Bendahhou S, Marionneau C, Haurogne K, Larroque MM, Derand R, Szuts V, Escande D, Demolombe S, Barhanin J. In vitro molecular interactions and distribution of KCNE family with KCNQ1 in the human heart. Cardiovasc Res 67: 529 –538, 2005. 56. Bryant SM, Wan X, Shipsey SJ, Hart G. Regional differences in the delayed rectifier current (IKr and IKs) contribute to the differences in action potential duration in basal left ventricular myocytes in guinea-pig. Cardiovasc Res 40: 322–331, 1998. 57. Burashnikov A, Antzelevitch C. Prominent I(Ks) in epicardium and endocardium contributes to development of transmural dispersion of repolarization but protects Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org 641 Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 20. Armoundas AA, Hobai IA, Tomaselli GF, Winslow RL, O’Rourke B. Role of sodiumcalcium exchanger in modulating the action potential of ventricular myocytes from normal and failing hearts. Circ Res 93: 46 –53, 2003. 39. Berkefeld H, Fakler B, Schulte U. Ca2⫹-activated K⫹ channels: from protein complexes to function. Physiol Rev 90: 1437–1459, 2010. SCHMITT, GRUNNET, AND OLESEN against development of early afterdepolarizations. J Cardiovasc Electrophysiol 13: 172– 177, 2002. 58. Burashnikov A, Antzelevitch C. Reinduction of atrial fibrillation immediately after termination of the arrhythmia is mediated by late phase 3 early afterdepolarizationinduced triggered activity. Circulation 107: 2355–2360, 2003. 59. Burashnikov A, Di Diego JM, Zygmunt AC, Belardinelli L, Antzelevitch C. Atriumselective sodium channel block as a strategy for suppression of atrial fibrillation: differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation 116: 1449 –1457, 2007. 59a.Burke MA, Mutharasan RK, Ardehali H. The sulfonylurea receptor, an atypical ATPbinding cassette protein, and its regulation of the KATP channel. Circ Res 102: 164 – 176, 2008. 60. Burstein B, Comtois P, Michael G, Nishida K, Villeneuve L, Yeh YH, Nattel S. Changes in connexin expression and the atrial fibrillation substrate in congestive heart failure. Circ Res 105: 1213–1222, 2009. 62. Calloe K, Cordeiro JM, Di Diego JM, Hansen RS, Grunnet M, Olesen SP, Antzelevitch C. A transient outward potassium current activator recapitulates the electrocardiographic manifestations of Brugada syndrome. Cardiovasc Res 81: 686 – 694, 2009. 63. Calloe K, Nof E, Jespersen T, Di Diego JM, Chlus N, Olesen SP, Antzelevitch C, Cordeiro JM. Comparison of the effects of a transient outward potassium channel activator on currents recorded from atrial and ventricular cardiomyocytes. J Cardiovasc Electrophysiol 22: 1057–1066, 2011. 64. Calloe K, Ravn LS, Schmitt N, Sui JL, Duno M, Haunso S, Grunnet M, Svendsen JH, Olesen SP. Characterizations of a loss-of-function mutation in the Kir3.4 channel subunit. Biochem Biophys Res Commun 364: 889 – 895, 2007. 65. Calloe K, Soltysinska E, Jespersen T, Lundby A, Antzelevitch C, Olesen SP, Cordeiro JM. Differential effects of the transient outward K⫹ current activator NS5806 in the canine left ventricle. J Mol Cell Cardiol 48: 191–200, 2010. 66. Camm AJ, Kirchhof P, Lip GYH, Schotten U, Savelieva I, Ernst S, Van Gelder IC, Al-Attar N, Hindricks G, Prendergast B, Heidbuchel H, Alfieri O, Angelini A, Atar D, Colonna P, De Caterina R, De Sutter J, Goette A, Gorenek B, Heldal M, Hohloser SH, Kolh P, Le Heuzey JY, Ponikowski P, Rutten FH. Guidelines for the management of atrial fibrillation: the Task Force for the Management of Atrial Fibrillation of the European Society of Cardiology (ESC). Europace 12: 1360 –1420, 2010. 67. Camm AJ, Lip GYH, De Caterina R, Savelieva I, Atar D, Hohnloser SH, Hindricks G, Kirchhof P. 2012 focused update of the ESC Guidelines for the management of atrial fibrillation: an update of the 2010 ESC Guidelines for the management of atrial fibrillation– developed with the special contribution of the European Heart Rhythm Association. Europace 14: 1385–1413, 2012. 68. Carlsson J, Miketic S, Windeler J, Cuneo A, Haun S, Micus S, Walter S, Tebbe U. Randomized trial of rate-control versus rhythm-control in persistent atrial fibrillation: the Strategies of Treatment of Atrial Fibrillation (STAF) study. J Am Coll Cardiol 41: 1690 –1696, 2003. 69. Carmeliet E. Voltage- and time-dependent block of the delayed K⫹ current in cardiac myocytes by dofetilide. J Pharmacol Exp Ther 262: 809 – 817, 1992. 70. Chang SH, Chang SN, Hwang JJ, Chiang FT, Tseng CD, Lee JK, Lai LP, Lin JL, Wu CK, Tsai CT. Significant association of rs13376333 in KCNN3 on chromosome 1q21 with atrial fibrillation in a Taiwanese population. Circ J 76: 184 –188, 2012. 75. Choisy SC, Arberry LA, Hancox JC, James AF. Increased susceptibility to atrial tachyarrhythmia in spontaneously hypertensive rat hearts. Hypertension 49: 498 –505, 2007. 76. Choy AM, Lang CC, Chomsky DM, Rayos GH, Wilson JR, Roden DM. Normalization of acquired QT prolongation in humans by intravenous potassium. Circulation 96: 2149 –2154, 1997. 77. Christ T, Boknik P, Wöhrl S, Wettwer E, Graf EM, Bosch RF, Knaut M, Schmitz W, Ravens U, Dobrev D. L-type Ca2⫹ current downregulation in chronic human atrial fibrillation is associated with increased activity of protein phosphatases. Circulation 110: 2651–2657, 2004. 78. Christ T, Wettwer E, Voigt N, Hala O, Radicke S, Matschke K, Varro A, Dobrev D, Ravens U. Pathology-specific effects of the IKur/Ito/IK,ACh blocker AVE0118 on ion channels in human chronic atrial fibrillation. Br J Pharmacol 154: 1619 –1630, 2008. 79. Christophersen IE, Olesen MS, Liang B, Andersen MN, Larsen AP, Nielsen JB, Haunsø S, Olesen SP, Tveit A, Svendsen JH, Schmitt N. Genetic variation in KCNA5: impact on the atrial-specific potassium current IKur in patients with lone atrial fibrillation. Eur Heart J 34: 1517–1525, 2013. 80. Colinas O, Gallego M, Setién R, López-López JR, Pérez-García MT, Casis O. Differential modulation of Kv4.2 and Kv43 channels by calmodulin-dependent protein kinase II in rat cardiac myocytes. Am J Physiol Heart Circ Physiol 291: H1978 –H1987, 2006. 81. Comtois P, Kneller J, Nattel S. Of circles and spirals: bridging the gap between the leading circle and spiral wave concepts of cardiac reentry. Europace 7 Suppl 2: 10 –20, 2005. 82. Conrath CE, Opthof T. Ventricular repolarization: an overview of (patho)physiology, sympathetic effects and genetic aspects. Prog Biophys Mol Biol 92: 269 –307, 2006. 83. Cooper MJ, Hunt LJ, Richards DA, Denniss AR, Uther JB, Ross DL. Effect of repetition of extrastimuli on sensitivity and reproducibility of mode of induction of ventricular tachycardia by programmed stimulation. J Am Coll Cardiol 11: 1260 –1267, 1988. 84. Cooper MJ. Assessment of efficacy of pharmacotherapy for ventricular tachycardia. Heart Lung Circ 16: 170 –175, 2007. 85. Cordeiro JM, Calloe K, Moise NS, Kornreich B, Giannandrea D, Di Diego JM, Olesen SP, Antzelevitch C. Physiological consequences of transient outward K⫹ current activation during heart failure in the canine left ventricle. J Mol Cell Cardiol 52: 1291– 1298, 2012. 86. Corley SD, Epstein AE, DiMarco JP, Domanski MJ, Geller N, Greene HL, Josephson RA, Kellen JC, Klein RC, Krahn AD, Mickel M, Mitchell LB, Nelson JD, Rosenberg Y, Schron E, Shemanski L, Waldo AL, Wyse DG. Relationships between sinus rhythm, treatment, and survival in the Atrial Fibrillation Follow-Up Investigation of Rhythm Management (AFFIRM) Study. Circulation 109: 1509 –1513, 2004. 87. Coronel R, Fiolet JW, Wilms-Schopman JG, Opthof T, Schaapherder AF, Janse MJ. Distribution of extracellular potassium and electrophysiologic changes during twostage coronary ligation in the isolated, perfused canine heart. Circulation 80: 165–177, 1989. 88. Coronel R, Wilders R, Verkerk AO, Wiegerinck RF, Benoist D, Bernus O. Electrophysiological changes in heart failure and their implications for arrhythmogenesis. Biochim Biophys Acta 1832: 2432–2441, 2013. 71. Chang SL, Tai CT, Lin YJ, Wongcharoen W, Lo LW, Lee KT, Chang SH, Tuan TC, Chen YJ, Hsieh MH, Tsao HM, Wu MH, Sheu MH, Chang CY, Chen SA. The role of left atrial muscular bundles in catheter ablation of atrial fibrillation. J Am Coll Cardiol 50: 964 –973, 2007. 89. Costa J, Lopes CM, Barsheshet A, Moss AJ, Migdalovich D, Ouellet G, McNitt S, Polonsky S, Robinson JL, Zareba W, Ackerman MJ, Benhorin J, Kaufman ES, Platonov PG, Shimizu W, Towbin JA, Vincent GM, Wilde AAM, Goldenberg I. Combined assessment of sex- and mutation-specific information for risk stratification in type 1 long QT syndrome. Heart Rhythm 9: 892– 898, 2012. 72. Abi-Char J, El-Haou S, Balse E, Neyroud N, Vranckx R, Coulombe A, Hatem SN. The anchoring protein SAP97 retains Kv1.5 channels in the plasma membrane of cardiac myocytes. Am J Physiol Heart Circ Physiol 294: H1851–H1861, 2008. 90. Courtemanche M, Ramirez RJ, Nattel S. Ionic targets for drug therapy and atrial fibrillation-induced electrical remodeling: insights from a mathematical model. Cardiovasc Res 42: 477– 489, 1999. 73. Chen X, Piacentino 3rd V, Furukawa S, Goldman B, Margulies KB, Houser SR. L-type Ca2⫹ channel density and regulation are altered in failing human ventricular myocytes and recover after support with mechanical assist devices. Circ Res 91: 517–524, 2002. 91. Courtemanche M, Winfree AT. Re-entrant rotating waves in a Beeler-Reuter based model of two-dimensional cardiac electrical activity. Int J Bifurc Chaos 1: 431– 444, 1991. 642 Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 61. Burstein B, Nattel S. Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillation. J Am Coll Cardiol 51: 802– 809, 2008. 74. Cherry EM, Fenton FH. Suppression of alternans and conduction blocks despite steep APD restitution: electrotonic, memory, and conduction velocity restitution effects. Am J Physiol Heart Circ Physiol 286: H2332–H2341, 2004. CARDIAC POTASSIUM CHANNELS AND ARRHYTHMIA 92. Cytrynbaum E, Keener JP. Stability conditions for the traveling pulse: modifying the restitution hypothesis. Chaos Woodbury N 12: 788 –799, 2002. 93. Davidenko JM, Pertsov AV, Salomonsz R, Baxter W, Jalife J. Stationary and drifting spiral waves of excitation in isolated cardiac muscle. Nature 355: 349 –351, 1992. 94. Decher N, Maier M, Dittrich W, Gassenhuber J, Brüggemann A, Busch AE, Steinmeyer K. Characterization of TASK-4, a novel member of the pH-sensitive, two-pore domain potassium channel family. FEBS Lett 492: 84 – 89, 2001. 95. Decher N, Wemhöner K, Rinné S, Netter MF, Zuzarte M, Aller MI, Kaufmann SG, Li XT, Meuth SG, Daut J, Sachse FB, Maier SKG. Knock-out of the potassium channel TASK-1 leads to a prolonged QT interval and a disturbed QRS complex. Cell Physiol Biochem 28: 77– 86, 2011. 96. Delpón E, Cordeiro JM, Núñez L, Thomsen PEB, Guerchicoff A, Pollevick GD, Wu Y, Kanters JK, Larsen CT, Hofman-Bang J, Burashnikov E, Christiansen M, Antzelevitch C. Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ Arrhythm Electrophysiol 1: 209 –218, 2008. 98. Dhamoon AS, Jalife J. The inward rectifier current (IK1) controls cardiac excitability and is involved in arrhythmogenesis. Heart Rhythm 2: 316 –324, 2005. 99. Dhar Malhotra J, Chen C, Rivolta I, Abriel H, Malhotra R, Mattei LN, Brosius FC, Kass RS, Isom LL. Characterization of sodium channel alpha- and beta-subunits in rat and mouse cardiac myocytes. Circulation 103: 1303–1310, 2001. 100. Diness JG, Hansen RS, Nissen JD, Jespersen T, Grunnet M. Antiarrhythmic effect of IKr activation in a cellular model of LQT3. Heart Rhythm 6: 100 –106, 2009. 101. Diness JG, Skibsbye L, Jespersen T, Bartels ED, Sørensen US, Hansen RS, Grunnet M. Effects on atrial fibrillation in aged hypertensive rats by Ca2⫹-activated K⫹ channel inhibition. Hypertension 57: 1129 –1135, 2011. 102. Diness JG, Sorensen US, Nissen JD, Al-Shahib B, Jespersen T, Grunnet M, Hansen RS. Inhibition of small-conductance Ca2⫹-activated K⫹ channels terminates and protects against atrial fibrillation. Circ Arrhythm Electrophysiol 3: 380 –390, 2010. 103. Diness TG, Yeh YH, Qi XY, Chartier D, Tsuji Y, Hansen RS, Olesen SP, Grunnet M, Nattel S. Antiarrhythmic properties of a rapid delayed-rectifier current activator in rabbit models of acquired long QT syndrome. Cardiovasc Res 79: 61– 69, 2008. 104. Dobrev D, Friedrich A, Voigt N, Jost N, Wettwer E, Christ T, Knaut M, Ravens U. The G protein-gated potassium current I(K,ACh) is constitutively active in patients with chronic atrial fibrillation. Circulation 112: 3697–3706, 2005. 105. Dobrev D, Ravens U. Remodeling of cardiomyocyte ion channels in human atrial fibrillation. Basic Res Cardiol 98: 137–148, 2003. 106. Dobrev D, Voigt N, Wehrens XHT. The ryanodine receptor channel as a molecular motif in atrial fibrillation: pathophysiological and therapeutic implications. Cardiovasc Res 89: 734 –743, 2011. 107. Donner BC, Schullenberg M, Geduldig N, Huning A, Mersmann J, Zacharowski K, Kovacevic A, Decking U, Aller MI, Schmidt KG. Functional role of TASK-1 in the heart: studies in TASK-1-deficient mice show prolonged cardiac repolarization and reduced heart rate variability. Basic Res Cardiol 106: 75– 87, 2011. 108. Dorian P. Mechanisms of action of class III agents and their clinical relevance. Europace 1 Suppl C: C6 –9, 2000. 109. Duprat F, Lesage F, Fink M, Reyes R, Heurteaux C, Lazdunski M. TASK, a human background K⫹ channel to sense external pH variations near physiological pH. EMBO J 16: 5464 –5471, 1997. 110. Echt DS, Liebson PR, Mitchell LB, Peters RW, Obias-Manno D, Barker AH, Arensberg D, Baker A, Friedman L, Greene HL. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial. N Engl J Med 324: 781–788, 1991. 111. Ehrlich JR, Cha TJ, Zhang L, Chartier D, Melnyk P, Hohnloser SH, Nattel S. Cellular electrophysiology of canine pulmonary vein cardiomyocytes: action potential and ionic current properties. J Physiol 551: 801– 813, 2003. 113. Ehrlich JR. Inward rectifier potassium currents as a target for atrial fibrillation therapy. J Cardiovasc Pharmacol 52: 129 –135, 2008. 114. Eijgelsheim M, Newton-Cheh C, Aarnoudse ALHJ, van Noord C, Witteman JCM, Hofman A, Uitterlinden AG, Stricker BHC. Genetic variation in NOS1AP is associated with sudden cardiac death: evidence from the Rotterdam Study. Hum Mol Genet 18: 4213– 4218, 2009. 115. Eisner DA, Vaughan-Jones RD. Do calcium-activated potassium channels exist in the heart? Cell Calcium 4: 371–386, 1983. 116. Eldstrom J, Fedida D. The voltage-gated channel accessory protein KCNE2: multiple ion channel partners, multiple ways to long QT syndrome. Expert Rev Mol Med 13: e38, 2011. 117. Ellinghaus P, Scheubel RJ, Dobrev D, Ravens U, Holtz J, Huetter J, Nielsch U, Morawietz H. Comparing the global mRNA expression profile of human atrial and ventricular myocardium with high-density oligonucleotide arrays. J Thorac Cardiovasc Surg 129: 1383–1390, 2005. 118. Ellinor PT, Lunetta KL, Glazer NL, Pfeufer A, Alonso A, Chung MK, Sinner MF, de Bakker PI, Mueller M, Lubitz SA, Fox E, Darbar D, Smith NL, Smith JD, Schnabel RB, Soliman EZ, Rice KM, Van Wagoner DR, Beckmann BM, van NC, Wang K, Ehret GB, Rotter JI, Hazen SL, Steinbeck G, Smith AV, Launer LJ, Harris TB, Makino S, Nelis M, Milan DJ, Perz S, Esko T, Kottgen A, Moebus S, Newton-Cheh C, Li M, Mohlenkamp S, Wang TJ, Kao WH, Vasan RS, Nothen MM, MacRae CA, Stricker BH, Hofman A, Uitterlinden AG, Levy D, Boerwinkle E, Metspalu A, Topol EJ, Chakravarti A, Gudnason V, Psaty BM, Roden DM, Meitinger T, Wichmann HE, Witteman JC, Barnard J, Arking DE, Benjamin EJ, Heckbert SR, Kaab S. Common variants in KCNN3 are associated with lone atrial fibrillation. Nat Genet 42: 240 –244, 2010. 119. Epstein AE, Bigger JT, Wyse DG, Romhilt DW, Reynolds-Haertle RA, Hallstrom AP. Events in the Cardiac Arrhythmia Suppression Trial (CAST): mortality in the entire population enrolled. J Am Coll Cardiol 18: 14 –19, 1991. 119a.Exome Variant Server. NHLBI Exome Sequencing Project (E.S.P.) (http://evs.gs.washington.edu/EVS/) Online 2012. 120. Fakler B, Brändle U, Glowatzki E, Weidemann S, Zenner HP, Ruppersberg JP. Strong voltage-dependent inward rectification of inward rectifier K⫹ channels is caused by intracellular spermine. Cell 80: 149 –154, 1995. 121. Fedida D. Vernakalant (RSD1235): a novel, atrial-selective antifibrillatory agent. Expert Opin Investig Drugs 16: 519 –532, 2007. 122. Feng J, Xu D, Wang Z, Nattel S. Ultrarapid delayed rectifier current inactivation in human atrial myocytes: properties and consequences. Am J Physiol Heart Circ Physiol 275: H1717–H1725, 1998. 123. Fenton FH, Cherry EM, Hastings HM, Evans SJ. Multiple mechanisms of spiral wave breakup in a model of cardiac electrical activity. Chaos Woodbury N 12: 852– 892, 2002. 124. Finley MR, Li Y, Hua F, Lillich J, Mitchell KE, Ganta S, Gilmour RF Jr, Freeman LC. Expression and coassociation of ERG1, KCNQ1, and KCNE1 potassium channel proteins in horse heart. Am J Physiol Heart Circ Physiol 283: H126 –H138, 2002. 124a.Flagg TP, Kurata HT, Masia R, Caputa G, Magnuson MA, Lefer DJ, Coetzee WA, Nichols CG. Differential structure of atrial and ventricular KATP: atrial KATP channels require SUR1. Circ Res 103: 1458 –1465, 2008. 125. Ford JW, Milnes JT. New drugs targeting the cardiac ultra-rapid delayed-rectifier current (I Kur): rationale, pharmacology and evidence for potential therapeutic value. J Cardiovasc Pharmacol 52: 105–120, 2008. 126. Fozzard HA, Hanck DA. Structure and function of voltage-dependent sodium channels: comparison of brain II and cardiac isoforms. Physiol Rev 76: 887–926, 1996. 127. Friedrich C. Abstracts of the 78th Annual Meeting of the German Society for Cardiology-Heart and Circulation Research. April 11–14, 2012 Mannheim, Germany. Clin Res Cardiol 101 Suppl 1: P984, 2012. 128. Fuster V, Ryden LE, Asinger RW, Cannom DS, Crijns HJ, Frye RL, Halperin JL, Kay GN, Klein WW, Levy S, McNamara RL, Prystowsky EN, Wann LS, Wyse DG. ACC/ AHA/ESC guidelines for the management of patients with atrial fibrillation. A report of Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org 643 Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 97. DeMaria CD, Soong TW, Alseikhan BA, Alvania RS, Yue DT. Calmodulin bifurcates the local Ca2⫹ signal that modulates P/Q-type Ca2⫹ channels. Nature 411: 484 – 489, 2001. 112. Ehrlich JR, Nattel S, Hohnloser SH. Atrial fibrillation and congestive heart failure: specific considerations at the intersection of two common and important cardiac disease sets. J Cardiovasc Electrophysiol 13: 399 – 405, 2002. SCHMITT, GRUNNET, AND OLESEN the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines and Policy Conferences (Committee to develop guidelines for the management of patients with atrial fibrillation) developed in collaboration with the North American Society of Pacing and Electrophysiology. Eur Heart J 22: 1852–1923, 2001. 129. Fuster V, Ryden LE, Cannom DS, Crijns HJ, Curtis AB, Ellenbogen KA, Halperin JL, Kay GN, Le Huezey JY, Lowe JE, Olsson SB, Prystowsky EN, Tamargo JL, Wann LS. 2011 ACCF/AHA/HRS Focused Updates Incorporated Into the ACC/AHA/ESC 2006 Guidelines for the Management of Patients with Atrial Fibrillation: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines Developed in partnership with the European Society of Cardiology and in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. J Am Coll Cardiol 57: e101– e198, 2011. 130. Fye WB. Tracing atrial fibrillation–100 years. N Engl J Med 355: 1412–1414, 2006. 131. Gaborit N, Le BS, Szuts V, Varro A, Escande D, Nattel S, Demolombe S. Regional and tissue specific transcript signatures of ion channel genes in the non-diseased human heart. J Physiol 582: 675– 693, 2007. 147. Gong Q, Anderson CL, January CT, Zhou Z. Role of glycosylation in cell surface expression and stability of HERG potassium channels. Am J Physiol Heart Circ Physiol 283: H77–H84, 2002. 148. Gong Q, Stump MR, Zhou Z. Inhibition of nonsense-mediated mRNA decay by antisense morpholino oligonucleotides restores functional expression of hERG nonsense and frameshift mutations in long-QT syndrome. J Mol Cell Cardiol 50: 223–229, 2011. 149. Gong Q, Zhang L, Vincent GM, Horne BD, Zhou Z. Nonsense mutations in hERG cause a decrease in mutant mRNA transcripts by nonsense-mediated mRNA decay in human long-QT syndrome. Circulation 116: 17–24, 2007. 150. González de la Fuente M, Barana A, Gómez R, Amorós I, Dolz-Gaitón P, Sacristán S, Atienza F, Pita A, Pinto Á, Fernández-Avilés F, Caballero R, Tamargo J, Delpón E. Chronic atrial fibrillation up-regulates 1-Adrenoceptors affecting repolarizing currents and action potential duration. Cardiovasc Res 97: 379 –388, 2013. 151. Gordon E, Lozinskaya IM, Lin Z, Semus SF, Blaney FE, Willette RN, Xu X. 2-[2-(3,4dichloro-phenyl)-2,3-dihydro-1H-isoindol-5-ylamino]-nicotinic acid (PD-307243) causes instantaneous current through human ether-a-go-go-related gene potassium channels. Mol Pharmacol 73: 639 – 651, 2008. 133. Gaborit N, Varro A, Le Bouter S, Szuts V, Escande D, Nattel S, Demolombe S. Gender-related differences in ion-channel and transporter subunit expression in nondiseased human hearts. J Mol Cell Cardiol 49: 639 – 646, 2010. 152. Gowd BMP, Thompson PD. Effect of female sex on cardiac arrhythmias. Cardiol Rev 20: 297–303, 2012. 134. Garfinkel A, Kim YH, Voroshilovsky O, Qu Z, Kil JR, Lee MH, Karagueuzian HS, Weiss JN, Chen PS. Preventing ventricular fibrillation by flattening cardiac restitution. Proc Natl Acad Sci USA 97: 6061– 6066, 2000. 153. Grunnet M, Bentzen BH, Sørensen US, Diness JG. Cardiac ion channels and mechanisms for protection against atrial fibrillation. Rev Physiol Biochem Pharmacol 162: 1–58, 2012. 135. Gaspo R, Bosch RF, Bou-Abboud E, Nattel S. Tachycardia-induced changes in Na⫹ current in a chronic dog model of atrial fibrillation. Circ Res 81: 1045–1052, 1997. 154. Grunnet M, Diness TG, Hansen RS, Olesen SP. Biophysical characterization of the short QT mutation hERG-N588K reveals a mixed gain-and loss-of-function. Cell Physiol Biochem 22: 611– 624, 2008. 136. Gauldie J, Hanson JM, Rumjanek FD, Shipolini RA, Vernon CA. The peptide components of bee venom. Eur J Biochem 61: 369 –376, 1976. 137. Van Gelder IC, Hagens VE, Bosker HA, Kingma JH, Kamp O, Kingma T, Said SA, Darmanata JI, Timmermans AJ, Tijssen JG, Crijns HJ. A comparison of rate control and rhythm control in patients with recurrent persistent atrial fibrillation. N Engl J Med 347: 1834 –1840, 2002. 138. Gentles RG, Grant-Young K, Hu S, Huang Y, Poss MA, Andres C, Fiedler T, Knox R, Lodge N, Weaver CD, Harden DG. Initial SAR studies on apamin-displacing 2-aminothiazole blockers of calcium-activated small conductance potassium channels. Bioorg Med Chem Lett 18: 5316 –5319, 2008. 139. Gierten J, Ficker E, Bloehs R, Schweizer PA, Zitron E, Scholz E, Karle C, Katus HA, Thomas D. The human cardiac K2P3.1 (TASK-1) potassium leak channel is a molecular target for the class III antiarrhythmic drug amiodarone. Naunyn-Schmiedebergs Arch Pharmacol 381: 261–270, 2010. 140. Girmatsion Z, Biliczki P, Bonauer A, Wimmer-Greinecker G, Scherer M, Moritz A, Bukowska A, Goette A, Nattel S, Hohnloser SH, Ehrlich JR. Changes in microRNA-1 expression and IK1 up-regulation in human atrial fibrillation. Heart Rhythm 6: 1802– 1809, 2009. 141. Giudicessi JR, Ackerman MJ. Determinants of incomplete penetrance and variable expressivity in heritable cardiac arrhythmia syndromes. Transl Res J Lab Clin Med 161: 1–14, 2013. 142. Gogelein H, Brendel J, Steinmeyer K, Strubing C, Picard N, Rampe D, Kopp K, Busch AE, Bleich M. Effects of the atrial antiarrhythmic drug AVE0118 on cardiac ion channels. Naunyn-Schmiedebergs Arch Pharmacol 370: 183–192, 2004. 143. Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell 128: 635– 638, 2007. 144. Goldoni D, Yarham JM, McGahon MK, O’Connor A, Guduric-Fuchs J, Edgar K, McDonald DM, Simpson DA, Collins A. A novel dual-fluorescence strategy for functionally validating microRNA targets in 3’ untranslated regions: regulation of the inward rectifier potassium channel K(ir)2.1 by miR-212. Biochem J 448: 103–113, 2012. 145. Goldstein SAN, Bayliss DA, Kim D, Lesage F, Plant LD, Rajan S. International Union of Pharmacology. LV. Nomenclature and molecular relationships of two-P potassium channels. Pharmacol Rev 57: 527–540, 2005. 644 155. Grunnet M, Hansen RS, Olesen SP. hERG1 channel activators: a new anti-arrhythmic principle. Prog Biophys Mol Biol 98: 347–362, 2008. 156. Grunnet M, Jensen BS, Olesen SP, Klaerke DA. Apamin interacts with all subtypes of cloned small-conductance Ca2⫹-activated K⫹ channels. Pflügers Arch Eur J Physiol 441: 544 –550, 2001. 157. Grunnet M, Jespersen T, Angelo K, Frøkjaer-Jensen C, Klaerke DA, Olesen SP, Jensen BS. Pharmacological modulation of SK3 channels. Neuropharmacology 40: 879 – 887, 2001. 158. Grunnet M, Jespersen T, Rasmussen HB, Ljungstrøm T, Jorgensen NK, Olesen SP, Klaerke DA. KCNE4 is an inhibitory subunit to the KCNQ1 channel. J Physiol 542: 119 –130, 2002. 159. Grunnet M, Kaufmann WA. Coassembly of big conductance Ca2⫹-activated K⫹ channels and L-type voltage-gated Ca2⫹ channels in rat brain. J Biol Chem 279: 36445– 36453, 2004. 160. Grunnet M, Olesen SP, Klaerke DA, Jespersen T. hKCNE4 inhibits the hKCNQ1 potassium current without affecting the activation kinetics. Biochem Biophys Res Commun 328: 1146 –1153, 2005. 161. Grunnet M, Strøbæk D, Olesen SP, Christophersen P. KCa-KCa5 families. In: Ion Channels: From Structure to Function. New York: Oxford Univ. Press, 2009, p. 403– 424. 162. Grunnet M. Repolarization of the cardiac action potential. Does an increase in repolarization capacity constitute a new anti-arrhythmic principle? Acta Physiol Oxf Engl 198 Suppl 676: 1– 48, 2010. 163. Guillemare E, Marion A, Nisato D, Gautier P. Inhibitory effects of dronedarone on muscarinic K⫹ current in guinea pig atrial cells. J Cardiovasc Pharmacol 36: 802– 805, 2000. 164. Guo D, Lu Z. Mechanism of IRK1 channel block by intracellular polyamines. J Gen Physiol 115: 799 – 814, 2000. 165. Guo W, Li H, Aimond F, Johns DC, Rhodes KJ, Trimmer JS, Nerbonne JM. Role of heteromultimers in the generation of myocardial transient outward K⫹ currents. Circ Res 90: 586 –593, 2002. Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 132. Gaborit N, Steenman M, Lamirault G, Le Meur N, Le Bouter S, Lande G, Léger J, Charpentier F, Christ T, Dobrev D, Escande D, Nattel S, Demolombe S. Human atrial ion channel and transporter subunit gene-expression remodeling associated with valvular heart disease and atrial fibrillation. Circulation 112: 471– 481, 2005. 146. Gómez R, Caballero R, Barana A, Amorós I, Calvo E, López JA, Klein H, Vaquero M, Osuna L, Atienza F, Almendral J, Pinto A, Tamargo J, Delpón E. Nitric oxide increases cardiac IK1 by nitrosylation of cysteine 76 of Kir2.1 channels. Circ Res 105: 383–392, 2009. CARDIAC POTASSIUM CHANNELS AND ARRHYTHMIA 166. Gurney A, Manoury B. Two-pore potassium channels in the cardiovascular system. Eur Biophys J 38: 305–318, 2009. 167. Haissaguerre M, Jais P, Shah DC, Takahashi A, Hocini M, Quiniou G, Garrigue S, Le MA, Le MP, Clementy J. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med 339: 659 – 666, 1998. 168. Hansen RS, Diness TG, Christ T, Demnitz J, Ravens U, Olesen SP, Grunnet M. Activation of human ether-a-go-go-related gene potassium channels by the diphenylurea 1,3-bis-(2-hydroxy-5-trifluoromethyl-phenyl)-urea (NS1643). Mol Pharmacol 69: 266 –277, 2006. 169. Hansen RS, Diness TG, Christ T, Wettwer E, Ravens U, Olesen SP, Grunnet M. Biophysical characterization of the new human ether-a-go-go-related gene channel opener NS3623 [N-(4-bromo-2-(1H-tetrazol-5-yl)-phenyl)-N’-(3’-trifluoromethylphenyl)urea]. Mol Pharmacol 70: 1319 –1329, 2006. 170. Hansen RS, Olesen SP, Grunnet M. Pharmacological activation of rapid delayed rectifier potassium current suppresses bradycardia-induced triggered activity in the isolated guinea pig heart. J Pharmacol Exp Ther 321: 996 –1002, 2007. 172. Hara M, Shvilkin A, Rosen MR, Danilo P Jr, Boyden PA. Steady-state and nonsteadystate action potentials in fibrillating canine atrium: abnormal rate adaptation and its possible mechanisms. Cardiovasc Res 42: 455– 469, 1999. 173. Harleton E, Besana A, Comas GM, Danilo P Jr, Rosen TS, Argenziano M, Rosen MR, Robinson RB, Feinmark SJ. Ability to induce atrial fibrillation in the peri-operative period is associated with phosphorylation-dependent inhibition of TWIK proteinrelated acid-sensitive potassium channel 1 (TASK-1). J Biol Chem 288: 2829 –2838, 2013. 174. Hedley PL, Jørgensen P, Schlamowitz S, Wangari R, Moolman-Smook J, Brink PA, Kanters JK, Corfield VA, Christiansen M. The genetic basis of long QT and short QT syndromes: a mutation update. Hum Mutat 30: 1486 –1511, 2009. 175. Heginbotham L, Lu Z, Abramson T, MacKinnon R. Mutations in the K⫹ channel signature sequence. Biophys J 66: 1061–1067, 1994. 176. Henry WL, Morganroth J, Pearlman AS, Clark CE, Redwood DR, Itscoitz SB, Epstein SE. Relation between echocardiographically determined left atrial size and atrial fibrillation. Circulation 53: 273–279, 1976. 177. Le Heuzey JY, De Ferrari GM, Radzik D, Santini M, Zhu J, Davy JM. A short-term, randomized, double-blind, parallel-group study to evaluate the efficacy and safety of dronedarone versus amiodarone in patients with persistent atrial fibrillation: the DIONYSOS study. J Cardiovasc Electrophysiol 21: 597– 605, 2010. 186. Hondeghem LM. Relative contributions of TRIaD and QT to proarrhythmia. J Cardiovasc Electrophysiol 18: 655– 657, 2007. 187. Hondeghem LM. QT prolongation is an unreliable predictor of ventricular arrhythmia. Heart Rhythm 5: 1210 –1212, 2008. 188. Ikeda T, Uchida T, Hough D, Lee JJ, Fishbein MC, Mandel WJ, Chen PS, Karagueuzian HS. Mechanism of spontaneous termination of functional reentry in isolated canine right atrium. Evidence for the presence of an excitable but nonexcited core. Circulation 94: 1962–1973, 1996. 189. Ionescu-Ittu R, Abrahamowicz M, Jackevicius CA, Essebag V, Eisenberg MJ, Wynant W, Richard H, Pilote L. Comparative effectiveness of rhythm control vs rate control drug treatment effect on mortality in patients with atrial fibrillation. Arch Intern Med 172: 997–1004, 2012. 189a.Isomoto S, Kurachi Y. Function, regulation, pharmacology, and molecular structure of ATP-sensitive K⫹ channels in the cardiovascular system. J Cardiovasc Electrophysiol 8: 1431–1446, 1997. 190. Iwasaki Y, Nishida K, Kato T, Nattel S. Atrial fibrillation pathophysiology: implications for management. Circulation 124: 2264 –2274, 2011. 191. Jager H, Adelman JP, Grissmer S. SK2 encodes the apamin-sensitive Ca2⫹-activated K⫹ channels in the human leukemic T cell line, Jurkat. FEBS Lett 469: 196 –202, 2000. 192. Janse MJ, Vermeulen JT, Opthof T, Coronel R, Wilms-Schopman FJ, Rademaker HM, Baartscheer A, Dekker LR. Arrhythmogenesis in heart failure. J Cardiovasc Electrophysiol 12: 496 – 499, 2001. 193. Janse MJ. Electrophysiological changes in heart failure and their relationship to arrhythmogenesis. Cardiovasc Res 61: 208 –217, 2004. 194. Jenkins DP, Strobaek D, Hougaard C, Jensen ML, Hummel R, Sorensen US, Christophersen P, Wulff H. Negative gating modulation by (R)-N-(benzimidazol-2-yl)1,2,3,4-tetrahydro-1-naphthylamine (NS8593) depends on residues in the inner pore vestibule: pharmacological evidence of deep-pore gating of KCa2 channels. Mol Pharmacol 79: 899 –909, 2011. 195. Jespersen T, Grunnet M, Olesen SP. The KCNQ1 potassium channel: from gene to physiological function. Physiology 20: 408 – 416, 2005. 196. Jespersen T, Membrez M, Nicolas CS, Pitard B, Staub O, Olesen SP, Baró I, Abriel H. The KCNQ1 potassium channel is down-regulated by ubiquitylating enzymes of the Nedd4/Nedd4-like family. Cardiovasc Res 74: 64 –74, 2007. 178. Hille B. G protein-coupled mechanisms and nervous signaling. Neuron 9: 187–195, 1992. 197. Jespersen T, Rasmussen HB, Grunnet M, Jensen HS, Angelo K, Dupuis DS, Vogel LK, Jorgensen NK, Klaerke DA, Olesen SP. Basolateral localisation of KCNQ1 potassium channels in MDCK cells: molecular identification of an N-terminal targeting motif. J Cell Sci 117: 4517– 4526, 2004. 179. Hohnloser SH, Connolly SJ, Crijns HJGM, Page RL, Seiz W, Torp-Petersen C. Rationale and design of ATHENA: a placebo-controlled, double-blind, parallel arm Trial to assess the efficacy of dronedarone 400 mg bid for the prevention of cardiovascular Hospitalization or death from any cause in patiENts with Atrial fibrillation/atrial flutter. J Cardiovasc Electrophysiol 19: 69 –73, 2008. 198. Jindal HK, Folco EJ, Liu GX, Koren G. Posttranslational modification of voltage-dependent potassium channel Kv1.5: COOH-terminal palmitoylation modulates its biological properties. Am J Physiol Heart Circ Physiol 294: H2012–H2021, 2008. 180. Hohnloser SH, Crijns HJGM, van Eickels M, Gaudin C, Page RL, Torp-Pedersen C, Connolly SJ. Effect of dronedarone on cardiovascular events in atrial fibrillation. N Engl J Med 360: 668 – 678, 2009. 181. Hohnloser SH, Kuck KH, Lilienthal J. Rhythm or rate control in atrial fibrillation– Pharmacological Intervention in Atrial Fibrillation (PIAF): a randomised trial. Lancet 356: 1789 –1794, 2000. 182. Holmes TC, Fadool DA, Ren R, Levitan IB. Association of Src tyrosine kinase with a human potassium channel mediated by SH3 domain. Science 274: 2089 –2091, 1996. 199. Johnson JN, Tester DJ, Perry J, Salisbury BA, Reed CR, Ackerman MJ. Prevalence of early-onset atrial fibrillation in congenital long QT syndrome. Heart Rhythm 5: 704 – 709, 2008. 200. Jones EMC, Roti Roti EC, Wang J, Delfosse SA, Robertson GA. Cardiac IKr channels minimally comprise hERG 1a and 1b subunits. J Biol Chem 279: 44690 – 44694, 2004. 201. Jonsson MKB, van der Heyden MAG, van Veen TAB. Deciphering hERG channels: molecular basis of the rapid component of the delayed rectifier potassium current. J Mol Cell Cardiol 53: 369 –374, 2012. 183. Homma N, Amran MS, Nagasawa Y, Hashimoto K. Topics on the Na⫹/Ca2⫹ exchanger: involvement of Na⫹/Ca2⫹ exchange system in cardiac triggered activity. J Pharmacol Sci 102: 17–21, 2006. 202. Kääb S, Dixon J, Duc J, Ashen D, Näbauer M, Beuckelmann DJ, Steinbeck G, McKinnon D, Tomaselli GF. Molecular basis of transient outward potassium current downregulation in human heart failure: a decrease in Kv4.3 mRNA correlates with a reduction in current density. Circulation 98: 1383–1393, 1998. 184. Hondeghem LM, Carlsson L, Duker G. Instability and triangulation of the action potential predict serious proarrhythmia, but action potential duration prolongation is antiarrhythmic. Circulation 103: 2004 –2013, 2001. 203. Kang J, Chen XL, Wang H, Ji J, Cheng H, Incardona J, Reynolds W, Viviani F, Tabart M, Rampe D. Discovery of a small molecule activator of the human ether-a-go-go-related gene (HERG) cardiac K⫹ channel. Mol Pharmacol 67: 827– 836, 2005. Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org 645 Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 171. Hansen RS, Olesen SP, Rønn LCB, Grunnet M. In vivo effects of the IKr agonist NS3623 on cardiac electrophysiology of the guinea pig. J Cardiovasc Pharmacol 52: 35– 41, 2008. 185. Hondeghem LM, Snyders DJ. Class III antiarrhythmic agents have a lot of potential but a long way to go. Reduced effectiveness and dangers of reverse use dependence. Circulation 81: 686 – 690, 1990. SCHMITT, GRUNNET, AND OLESEN 204. Kannel WB, Abbott RD, Savage DD, McNamara PM. Coronary heart disease and atrial fibrillation: the Framingham Study. Am Heart J 106: 389 –396, 1983. 205. Kao WHL, Arking DE, Post W, Rea TD, Sotoodehnia N, Prineas RJ, Bishe B, Doan BQ, Boerwinkle E, Psaty BM, Tomaselli GF, Coresh J, Siscovick DS, Marbán E, Spooner PM, Burke GL, Chakravarti A. Genetic variations in nitric oxide synthase 1 adaptor protein are associated with sudden cardiac death in US white community-based populations. Circulation 119: 940 –951, 2009. 206. Karagueuzian HS, Athill CA, Yashima M, Ikeda T, Wu TJ, Mandel WJ, Chen PS. Transmembrane potential properties of atrial cells at different sites of a spiral wave reentry: cellular evidence for an excitable but nonexcited core. Pacing Clin Electrophysiol 21: 2360 –2365, 1998. 207. Karma. Meandering transition in two-dimensional excitable media. Phys Rev Lett 65: 2824 –2827, 1990. 208. Kathöfer S, Röckl K, Zhang W, Thomas D, Katus H, Kiehn J, Kreye V, Schoels W, Karle C. Human beta(3)-adrenoreceptors couple to KvLQT1/MinK potassium channels in Xenopus oocytes via protein kinase C phosphorylation of the KvLQT1 protein. Naunyn-Schmiedebergs Arch Pharmacol 368: 119 –126, 2003. 210. Kaufmann SG, Westenbroek RE, Maass AH, Lange V, Renner A, Wischmeyer E, Bonz A, Muck J, Ertl G, Catterall WA, Scheuer T, Maier SKG. Distribution and function of sodium channel subtypes in human atrial myocardium. J Mol Cell Cardiol 61: 133–141, 2013. 211. Kawase A, Ikeda T, Nakazawa K, Ashihara T, Namba T, Kubota T, Sugi K, Hirai H. Widening of the excitable gap and enlargement of the core of reentry during atrial fibrillation with a pure sodium channel blocker in canine atria. Circulation 107: 905– 910, 2003. 212. Kennedy ME, Nemec J, Corey S, Wickman K, Clapham DE. GIRK4 confers appropriate processing and cell surface localization to G-protein-gated potassium channels. J Biol Chem 274: 2571–2582, 1999. 213. Kharche S, Garratt CJ, Boyett MR, Inada S, Holden AV, Hancox JC, Zhang H. Atrial proarrhythmia due to increased inward rectifier current [I(K1)] arising from KCNJ2 mutation–a simulation study. Prog Biophys Mol Biol 98: 186 –197, 2008. 214. Kiesecker C, Zitron E, Scherer D, Lueck S, Bloehs R, Scholz EP, Pirot M, Kathöfer S, Thomas D, Kreye VAW, Kiehn J, Borst MM, Katus HA, Schoels W, Karle CA. Regulation of cardiac inwardly rectifying potassium current IK1 and Kir2.x channels by endothelin-1. J Mol Med 84: 46 –56, 2006. 215. Kim D, Fujita A, Horio Y, Kurachi Y. Cloning and functional expression of a novel cardiac two-pore background K⫹ channel (cTBAK-1). Circ Res 82: 513–518, 1998. 216. Kim GH. MicroRNA regulation of cardiac conduction and arrhythmias. Transl Res J Lab Clin Med 161: 381–392, 2013. 217. Kisselbach J, Schweizer PA, Gerstberger R, Becker R, Katus HA, Thomas D. Enhancement of K2P2.1 (TREK1) background currents expressed in Xenopus oocytes by voltage-gated K⫹ channel  subunits. Life Sci 91: 377–383, 2012. 218. Kjekshus J. Arrhythmias and mortality in congestive heart failure. Am J Cardiol 65: 42I– 48I, 1990. 224. Kostin S, Klein G, Szalay Z, Hein S, Bauer EP, Schaper J. Structural correlate of atrial fibrillation in human patients. Cardiovasc Res 54: 361–379, 2002. 225. Krzystanek K, Rasmussen HB, Grunnet M, Staub O, Olesen SP, Abriel H, Jespersen T. Deubiquitylating enzyme USP2 counteracts Nedd4 –2-mediated downregulation of KCNQ1 potassium channels. Heart Rhythm 9: 440 – 448, 2012. 226. Kubo Y, Baldwin TJ, Jan YN, Jan LY. Primary structure and functional expression of a mouse inward rectifier potassium channel. Nature 362: 127–133, 1993. 227. Kubota T, Shimizu W, Kamakura S, Horie M. Hypokalemia-induced long QT syndrome with an underlying novel missense mutation in S4 –S5 linker of KCNQ1. J Cardiovasc Electrophysiol 11: 1048 –1054, 2000. 228. Kupershmidt S, Snyders DJ, Raes A, Roden DM. A K⫹ channel splice variant common in human heart lacks a C-terminal domain required for expression of rapidly activating delayed rectifier current. J Biol Chem 273: 27231–27235, 1998. 229. Lafrenière RG, Cader MZ, Poulin JF, Andres-Enguix I, Simoneau M, Gupta N, Boisvert K, Lafrenière F, McLaughlan S, Dubé MP, Marcinkiewicz MM, Ramagopalan S, Ansorge O, Brais B, Sequeiros J, Pereira-Monteiro JM, Griffiths LR, Tucker SJ, Ebers G, Rouleau GA. A dominant-negative mutation in the TRESK potassium channel is linked to familial migraine with aura. Nat Med 16: 1157–1160, 2010. 230. Lagrutta A, Wang J, Fermini B, Salata JJ. Novel, potent inhibitors of human Kv1.5 K⫹ channels and ultrarapidly activating delayed rectifier potassium current. J Pharmacol Exp Ther 317: 1054 –1063, 2006. 231. Larsen AP, Bentzen BH, Grunnet M. Differential effects of Kv11.1 activators on Kv111a, Kv111b and Kv111a/Kv111b channels. Br J Pharmacol 161: 614 – 628, 2010. 232. Larsen AP, Olesen SP, Grunnet M, Jespersen T. Characterization of hERG1a and hERG1b potassium channels–a possible role for hERG1b in the I(Kr) current. Pflügers Arch 456: 1137–1148, 2008. 233. Larsen AP. Role of ERG1 isoforms in modulation of ERG1 channel trafficking and function. Pflügers Arch 460: 803– 812, 2010. 234. Lee HC, Rudy Y, Po-Yuan P, Sheu SH, Chang JG, Cui J. Modulation of KCNQ1 alternative splicing regulates cardiac IKs and action potential repolarization. Heart Rhythm 10: 1220 –1228, 2013. 235. Lee KS. Ibutilide, a new compound with potent class III antiarrhythmic activity, activates a slow inward Na⫹ current in guinea pig ventricular cells. J Pharmacol Exp Ther 262: 99 –108, 1992. 236. Leonoudakis D, Mailliard W, Wingerd K, Clegg D, Vandenberg C. Inward rectifier potassium channel Kir2.2 is associated with synapse-associated protein SAP97. J Cell Sci 114: 987–998, 2001. 237. Lesage F, Lazdunski M. Molecular and functional properties of two-pore-domain potassium channels. Am J Physiol Renal Physiol 279: F793–F801, 2000. 238. Levin MD, Lu MM, Petrenko NB, Hawkins BJ, Gupta TH, Lang D, Buckley PT, Jochems J, Liu F, Spurney CF, Yuan LJ, Jacobson JT, Brown CB, Huang L, Beermann F, Margulies KB, Madesh M, Eberwine JH, Epstein JA, Patel VV. Melanocyte-like cells in the heart and pulmonary veins contribute to atrial arrhythmia triggers. J Clin Invest 119: 3420 –3436, 2009. 219. Kneller J, Kalifa J, Zou R, Zaitsev AV, Warren M, Berenfeld O, Vigmond EJ, Leon LJ, Nattel S, Jalife J. Mechanisms of atrial fibrillation termination by pure sodium channel blockade in an ionically-realistic mathematical model. Circ Res 96: e35– 47, 2005. 239. Li D, Fareh S, Leung TK, Nattel S. Promotion of atrial fibrillation by heart failure in dogs: atrial remodeling of a different sort. Circulation 100: 87–95, 1999. 220. Kneller J, Sun H, Leblanc N, Nattel S. Remodeling of Ca2⫹-handling by atrial tachycardia: evidence for a role in loss of rate-adaptation. Cardiovasc Res 54: 416 – 426, 2002. 240. Li D, Melnyk P, Feng J, Wang Z, Petrecca K, Shrier A, Nattel S. Effects of experimental heart failure on atrial cellular and ionic electrophysiology. Circulation 101: 2631–2638, 2000. 221. Kneller J, Zou R, Vigmond EJ, Wang Z, Leon LJ, Nattel S. Cholinergic atrial fibrillation in a computer model of a two-dimensional sheet of canine atrial cells with realistic ionic properties. Circ Res 90: E73– 87, 2002. 241. Li GR, Feng J, Yue L, Carrier M. Transmural heterogeneity of action potentials and Ito1 in myocytes isolated from the human right ventricle. Am J Physiol Heart Circ Physiol 275: H369 –H377, 1998. 222. Køber L, Torp-Pedersen C, McMurray JJV, Gøtzsche O, Lévy S, Crijns H, Amlie J, Carlsen J, Dronedarone Study Group. Increased mortality after dronedarone therapy for severe heart failure. N Engl J Med 358: 2678 –2687, 2008. 242. Li GR, Lau CP, Leung TK, Nattel S. Ionic current abnormalities associated with prolonged action potentials in cardiomyocytes from diseased human right ventricles. Heart Rhythm 1: 460 – 468, 2004. 646 Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 209. Kaufman ES, Zimmermann PA, Wang T, Dennish 3rd GW, Barrell PD, Chandler ML, Greene HL. Risk of proarrhythmic events in the Atrial Fibrillation Follow-up Investigation of Rhythm Management (AFFIRM) study: a multivariate analysis. J Am Coll Cardiol 44: 1276 –1282, 2004. 223. Kong W, Po S, Yamagishi T, Ashen MD, Stetten G, Tomaselli GF. Isolation and characterization of the human gene encoding Ito: further diversity by alternative mRNA splicing. Am J Physiol Heart Circ Physiol 275: H1963–H1970, 1998. CARDIAC POTASSIUM CHANNELS AND ARRHYTHMIA 243. Li GR, Lau CP, Shrier A. Heterogeneity of sodium current in atrial vs epicardial ventricular myocytes of adult guinea pig hearts. J Mol Cell Cardiol 34: 1185–1194, 2002. 244. Li ML, Li T, Lei M, Tan XQ, Yang Y, Liu TP, Pei J, Zeng XR. Increased small conductance calcium-activated potassium channel (SK2 channel) current in atrial myocytes of patients with persistent atrial fibrillation. Zhonghua Xin Xue Guan Bing Za Zhi 39: 147–151, 2011. 245. Li N, Timofeyev V, Tuteja D, Xu D, Lu L, Zhang Q, Zhang Z, Singapuri A, Albert TR, Rajagopal AV, Bond CT, Periasamy M, Adelman J, Chiamvimonvat N. Ablation of a Ca2⫹-activated K⫹ channel (SK2 channel) results in action potential prolongation in atrial myocytes and atrial fibrillation. J Physiol 587: 1087–1100, 2009. 246. Liang B, Nissen JD, Laursen M, Wang X, Skibsbye L, Hearing MC, Andersen MN, Rasmussen HB, Wickman K, Grunnet M, Olesen SP, Jespersen T. G-protein-coupled inward rectifier potassium current contributes to ventricular repolarization. Cardiovasc Res 101: 175–184, 2014. 262. Lucchesi BR, Mullane KM. Leukocytes and ischemia-induced myocardial injury. Annu Rev Pharmacol Toxicol 26: 201–224, 1986. 263. Lundby A, Jespersen T, Schmitt N, Grunnet M, Olesen SP, Cordeiro JM, Calloe K. Effect of the I(to) activator NS5806 on cloned K(V)4 channels depends on the accessory protein KChIP2. Br J Pharmacol 160: 2028 –2044, 2010. 264. Lundby A, Ravn LS, Svendsen JH, Hauns S, Olesen SP, Schmitt N. KCNE3 mutation V17M identified in a patient with lone atrial fibrillation. Cell Physiol Biochem 21: 47–54, 2008. 265. Lundby A, Ravn LS, Svendsen JH, Olesen SP, Schmitt N. KCNQ1 mutation Q147R is associated with atrial fibrillation and prolonged QT interval. Heart Rhythm 4: 1532– 1541, 2007. 266. Lundby A, Tseng GN, Schmitt N. Structural basis for K(V)7.1-KCNE(x) interactions in the I(Ks) channel complex. Heart Rhythm 7: 708 –713, 2010. 267. Luo X, Pan Z, Shan H, Xiao J, Sun X, Wang N, Lin H, Xiao L, Maguy A, Qi XY, Li Y, Gao X, Dong D, Zhang Y, Bai Y, Ai J, Sun L, Lu H, Luo XY, Wang Z, Lu Y, Yang B, Nattel S. MicroRNA-26 governs profibrillatory inward-rectifier potassium current changes in atrial fibrillation. J Clin Invest 123: 1939 –1951, 2013. 248. Liegeois JF, Mercier F, Graulich A, Graulich-Lorge F, Scuvee-Moreau J, Seutin V. Modulation of small conductance calcium-activated potassium (SK) channels: a new challenge in medicinal chemistry. Curr Med Chem 10: 625– 647, 2003. 268. Lüscher C, Slesinger PA. Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat Rev Neurosci 11: 301–315, 2010. 249. Light PE, Bladen C, Winkfein RJ, Walsh MP, French RJ. Molecular basis of protein kinase C-induced activation of ATP-sensitive potassium channels. Proc Natl Acad Sci USA 97: 9058 –9063, 2000. 269. Machida T, Hashimoto N, Kuwahara I, Ogino Y, Matsuura J, Yamamoto W, Itano Y, Zamma A, Matsumoto R, Kamon J, Kobayashi T, Ishiwata N, Yamashita T, Ogura T, Nakaya H. Effects of a highly selective acetylcholine-activated K⫹ channel blocker on experimental atrial fibrillation. Circ Arrhythm Electrophysiol 4: 94 –102, 2011. 250. Limberg SH, Netter MF, Rolfes C, Rinne S, Schlichthorl G, Zuzarte M, Vassiliou T, Moosdorf R, Wulf H, Daut J, Sachse FB, Decher N. TASK-1 channels may modulate action potential duration of human atrial cardiomyocytes. Cell Physiol Biochem 28: 613– 624, 2011. 270. Maingret F, Patel AJ, Lesage F, Lazdunski M, Honoré E. Mechano- or acid stimulation, two interactive modes of activation of the TREK-1 potassium channel. J Biol Chem 274: 26691–26696, 1999. 251. Liu DW, Antzelevitch C. Characteristics of the delayed rectifier current (IKr and IKs) in canine ventricular epicardial, midmyocardial, and endocardial myocytes. A weaker IKs contributes to the longer action potential of the M cell. Circ Res 76: 351–365, 1995. 252. Liu QH, Li XL, Xu YW, Lin YY, Cao JM, Wu BW. A novel discovery of IK1 channel agonist: zacopride selectively enhances IK1 current and suppresses triggered arrhythmias in the rat. J Cardiovasc Pharmacol 59: 37– 48, 2012. 253. Liu W, Saint DA. Heterogeneous expression of tandem-pore K⫹ channel genes in adult and embryonic rat heart quantified by real-time polymerase chain reaction. Clin Exp Pharmacol Physiol 31: 174 –178, 2004. 254. Lloyd-Jones DM, Wang TJ, Leip EP, Larson MG, Levy D, Vasan RS, D’Agostino RB, Massaro JM, Beiser A, Wolf PA, Benjamin EJ. Lifetime risk for development of atrial fibrillation: the Framingham Heart Study. Circulation 110: 1042–1046, 2004. 255. London B, Trudeau MC, Newton KP, Beyer AK, Copeland NG, Gilbert DJ, Jenkins NA, Satler CA, Robertson GA. Two isoforms of the mouse ether-a-go-go-related gene coassemble to form channels with properties similar to the rapidly activating component of the cardiac delayed rectifier K⫹ current. Circ Res 81: 870 – 878, 1997. 256. London B. Whither art thou, SCN10A, and what art thou doing? Circ Res 111: 268 – 270, 2012. 257. Long SB, Campbell EB, MacKinnon R. Crystal structure of a mammalian voltagedependent Shaker family K⫹ channel. Science 309: 897–903, 2005. 271. Mangoni ME, Couette B, Bourinet E, Platzer J, Reimer D, Striessnig J, Nargeot J. Functional role of L-type Cav1.3 Ca2⫹ channels in cardiac pacemaker activity. Proc Natl Acad Sci USA 100: 5543–5548, 2003. 272. Mao J, Wang X, Chen F, Wang R, Rojas A, Shi Y, Piao H, Jiang C. Molecular basis for the inhibition of G protein-coupled inward rectifier K⫹ channels by protein kinase C. Proc Natl Acad Sci USA 101: 1087–1092, 2004. 273. Marban E, O’Rourke B. Calcium channels: structure, function, regulation. In: Cardiac Electrophysiology. From Cell to Bedside. Philadelphia, PA: Saunders, 1995, p. 11–21. 274. Marroni F, Pfeufer A, Aulchenko YS, Franklin CS, Isaacs A, Pichler I, Wild SH, Oostra BA, Wright AF, Campbell H, Witteman JC, Kääb S, Hicks AA, Gyllensten U, Rudan I, Meitinger T, Pattaro C, van Duijn CM, Wilson JF, Pramstaller PP, Consortium EUROSPAN. A genome-wide association scan of RR and QT interval duration in 3 European genetically isolated populations: the EUROSPAN project. Circ Cardiovasc Genet 2: 322–328, 2009. 275. Marshall GE, Russell JA, Tellez JO, Jhund PS, Currie S, Dempster J, Boyett MR, Kane KA, Rankin AC, Workman AJ. Remodelling of human atrial K⫹ currents but not ion channel expression by chronic -blockade. Pflügers Arch 463: 537–548, 2012. 276. Marx SO, Kurokawa J, Reiken S, Motoike H, D’Armiento J, Marks AR, Kass RS. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science 295: 496 – 499, 2002. 258. Lopatin AN, Nichols CG. Inward rectifiers in the heart: an update on I(K1). J Mol Cell Cardiol 33: 625– 638, 2001. 277. Marzian S, Stansfeld PJ, Rapedius M, Rinné S, Nematian-Ardestani E, Abbruzzese JL, Steinmeyer K, Sansom MSP, Sanguinetti MC, Baukrowitz T, Decher N. Side pockets provide the basis for a new mechanism of Kv channel-specific inhibition. Nat Chem Biol 9: 507–513, 2013. 259. Lu HR, Vlaminckx E, Hermans AN, Rohrbacher J, Van Ammel K, Towart R, Pugsley M, Gallacher DJ. Predicting drug-induced changes in QT interval and arrhythmias: QTshortening drugs point to gaps in the ICHS7B Guidelines. Br J Pharmacol 154: 1427– 1438, 2008. 278. Matkovich SJ, Wang W, Tu Y, Eschenbacher WH, Dorn LE, Condorelli G, Diwan A, Nerbonne JM, Dorn GW 2nd. MicroRNA-133a protects against myocardial fibrosis and modulates electrical repolarization without affecting hypertrophy in pressureoverloaded adult hearts. Circ Res 106: 166 –175, 2010. 260. Lu L, Zhang Q, Timofeyev V, Zhang Z, Young JN, Shin HS, Knowlton AA, Chiamvimonvat N. Molecular coupling of a Ca2⫹-activated K⫹ channel to L-type Ca2⫹ channels via alpha-actinin2. Circ Res 100: 112–120, 2007. 279. McCrossan ZA, Abbott GW. The MinK-related peptides. Neuropharmacology 47: 787– 821, 2004. 261. Lu Z, Abe J, Taunton J, Lu Y, Shishido T, McClain C, Yan C, Xu SP, Spangenberg TM, Xu H. Reactive oxygen species-induced activation of p90 ribosomal S6 kinase pro- 280. McDonald TV, Yu Z, Ming Z, Palma E, Meyers MB, Wang KW, Goldstein SA, Fishman GI. A minK-HERG complex regulates the cardiac potassium current I(Kr). Nature 388: 289 –292, 1997. Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org 647 Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 247. Liang B, Soka M, Christensen AH, Olesen MS, Larsen AP, Knop FK, Wang F, Nielsen JB, Andersen MN, Humphreys D, Mann SA, Huttner IG, Vandenberg JI, Svendsen JH, Haunsø S, Preiss T, Seebohm G, Olesen SP, Schmitt N, Fatkin D. Genetic variation in the two-pore domain potassium channel, TASK-1, may contribute to an atrial substrate for arrhythmogenesis. J Mol Cell Cardiol doi:10.1016/j.yjmcc.2013.12.014. longs cardiac repolarization through inhibiting outward K⫹ channel activity. Circ Res 103: 269 –278, 2008. SCHMITT, GRUNNET, AND OLESEN 281. Medina I, Krapivinsky G, Arnold S, Kovoor P, Krapivinsky L, Clapham DE. A switch mechanism for G beta gamma activation of I(KACh). J Biol Chem 275: 29709 –29716, 2000. 300. Nattel S, Maguy A, Le Bouter S, Yeh YH. Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev 87: 425– 456, 2007. 282. Milan DJ, Lubitz SA, Kääb S, Ellinor PT. Genome-wide association studies in cardiac electrophysiology: recent discoveries and implications for clinical practice. Heart Rhythm 7: 1141–1148, 2010. 301. Nattel S, Shiroshita-Takeshita A, Brundel BJJM, Rivard L. Mechanisms of atrial fibrillation: lessons from animal models. Prog Cardiovasc Dis 48: 9 –28, 2005. 283. Miller AN, Long SB. Crystal structure of the human two-pore domain potassium channel K2P1. Science 335: 432– 436, 2012. 284. Milne JR, Hellestrand KJ, Bexton RS, Burnett PJ, Debbas NM, Camm AJ. Class 1 antiarrhythmic drugs– characteristic electrocardiographic differences when assessed by atrial and ventricular pacing. Eur Heart J 5: 99 –107, 1984. 285. Milstein ML, Musa H, Balbuena DP, Anumonwo JMB, Auerbach DS, Furspan PB, Hou L, Hu B, Schumacher SM, Vaidyanathan R, Martens JR, Jalife J. Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc Natl Acad Sci USA 109: E2134 –2143, 2012. 287. Mohammad-Panah R, Demolombe S, Neyroud N, Guicheney P, Kyndt F, van den Hoff M, Baró I, Escande D. Mutations in a dominant-negative isoform correlate with phenotype in inherited cardiac arrhythmias. Am J Hum Genet 64: 1015–1023, 1999. 288. Mori K, Hara Y, Saito T, Masuda Y, Nakaya H. Anticholinergic effects of class III antiarrhythmic drugs in guinea pig atrial cells. Different molecular mechanisms. Circulation 91: 2834 –2843, 1995. 289. Mort M, Ivanov D, Cooper DN, Chuzhanova NA. A meta-analysis of nonsense mutations causing human genetic disease. Hum Mutat 29: 1037–1047, 2008. 290. Moss AJ, Robinson JL, Gessman L, Gillespie R, Zareba W, Schwartz PJ, Vincent GM, Benhorin J, Heilbron EL, Towbin JA, Priori SG, Napolitano C, Zhang L, Medina A, Andrews ML, Timothy K. Comparison of clinical and genetic variables of cardiac events associated with loud noise versus swimming among subjects with the long QT syndrome. Am J Cardiol 84: 876 – 879, 1999. 291. Mullally J, Goldenberg I, Moss AJ, Lopes CM, Ackerman MJ, Zareba W, McNitt S, Robinson JL, Benhorin J, Kaufman ES, Towbin JA, Barsheshet A. Risk of life-threatening cardiac events among patients with long QT syndrome and multiple mutations. Heart Rhythm 10: 378 –382, 2013. 292. Murata M, Buckett PD, Zhou J, Brunner M, Folco E, Koren G. SAP97 interacts with Kv1.5 in heterologous expression systems Am J Physiol Heart Circ Physiol 281: H2575– H2584, 2001. 293. Näbauer M, Beuckelmann DJ, Uberfuhr P, Steinbeck G. Regional differences in current density and rate-dependent properties of the transient outward current in subepicardial and subendocardial myocytes of human left ventricle. Circulation 93: 168 – 177, 1996. 294. Nagaraj C, Tang B, Bálint Z, Wygrecka M, Hrzenjak A, Kwapiszewska G, Stacher E, Lindenmann J, Weir EK, Olschewski H, Olschewski A. Src tyrosine kinase is crucial for potassium channel function in human pulmonary arteries. Eur Respir J 41: 85–95, 2013. 295. Nagy N, Szuts V, Horvath Z, Seprenyi G, Farkas AS, Acsai K, Prorok J, Bitay M, Kun A, Pataricza J, Papp JG, Nanasi PP, Varro A, Toth A. Does small-conductance calciumactivated potassium channel contribute to cardiac repolarization? J Mol Cell Cardiol 47: 656 – 663, 2009. 296. Nakajima T, Wu J, Kaneko Y, Ashihara T, Ohno S, Irie T, Ding WG, Matsuura H, Kurabayashi M, Horie M. KCNE3 T4A as the genetic basis of Brugada-pattern electrocardiogram. Circ J 76: 2763–2772, 2012. 297. Namekata I, Tsuruoka N, Tsuneoka Y, Matsuda T, Takahara A, Tanaka Y, Suzuki T, Takahashi T, Iida-Tanaka N, Tanaka H. Blocking effect of NIP-142 on the KCNQ1/ KCNE1 channel current expressed in HEK293 cells. Biol Pharm Bull 34: 153–155, 2011. 298. Napolitano C, Wilson J. Gene connection of the heart [Online]. http://www.fsm. it/cardmoc/. 299. Nattel S, Burstein B, Dobrev D. Atrial remodeling and atrial fibrillation: mechanisms and implications. Circ Arrhythm Electrophysiol 1: 62–73, 2008. 648 303. Nattel S. New ideas about atrial fibrillation 50 years on. Nature 415: 219 –226, 2002. 304. Nattel S. Calcium-activated potassium current: a novel ion channel candidate in atrial fibrillation. J Physiol 587: 1385–1386, 2009. 305. Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev 85: 1205–1253, 2005. 306. Newton-Cheh C, Eijgelsheim M, Rice KM, de Bakker PIW, Yin X, Estrada K, Bis JC, Marciante K, Rivadeneira F, Noseworthy PA, Sotoodehnia N, Smith NL, Rotter JI, Kors JA, Witteman JCM, Hofman A, Heckbert SR, O’Donnell CJ, Uitterlinden AG, Psaty BM, Lumley T, Larson MG, Stricker BHC. Common variants at ten loci influence QT interval duration in the QTGEN Study. Nat Genet 41: 399 – 406, 2009. 307. Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, Faure S, Gary F, Coumel P, Petit C, Schwartz K, Guicheney P. A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat Genet 15: 186 –189, 1997. 308. Nielsen JB, Graff C, Pietersen A, Lind B, Struijk JJ, Olesen MS, Haunsø S, Gerds TA, Svendsen JH, Køber L, Holst AG. J-shaped association between QTc interval duration and the risk of atrial fibrillation: results from the Copenhagen ECG study. J Am Coll Cardiol 61: 2557–2564, 2013. 309. Nilius B, Hess P, Lansman JB, Tsien RW. A novel type of cardiac calcium channel in ventricular cells. Nature 316: 443– 446, 1985. 310. Nissen JD, Diness JG, Diness TG, Hansen RS, Grunnet M, Jespersen T. Pharmacologically induced long QT type 2 can be rescued by activation of IKs with benzodiazepine R-L3 in isolated guinea pig cardiomyocytes. J Cardiovasc Pharmacol 54: 169 –177, 2009. 311. Niwa N, Nerbonne JM. Molecular determinants of cardiac transient outward potassium current [I(to)] expression and regulation. J Mol Cell Cardiol 48: 12–25, 2010. 313. Núñez L, Vaquero M, Gómez R, Caballero R, Mateos-Cáceres P, Macaya C, Iriepa I, Gálvez E, López-Farré A, Tamargo J, Delpón E. Nitric oxide blocks hKv1.5 channels by S-nitrosylation and by a cyclic GMP-dependent mechanism. Cardiovasc Res 72: 80 – 89, 2006. 314. Nuss HB, Kääb S, Kass DA, Tomaselli GF, Marbán E. Cellular basis of ventricular arrhythmias and abnormal automaticity in heart failure. Am J Physiol Heart Circ Physiol 277: H80 –H91, 1999. 315. O’Connell AD, Morton MJ, Hunter M. Two-pore domain K⫹ channels-molecular sensors. Biochim Biophys Acta 1566: 152–161, 2002. 316. Ohno S, Toyoda F, Zankov DP, Yoshida H, Makiyama T, Tsuji K, Honda T, Obayashi K, Ueyama H, Shimizu W, Miyamoto Y, Kamakura S, Matsuura H, Kita T, Horie M. Novel KCNE3 mutation reduces repolarizing potassium current and associated with long QT syndrome. Hum Mutat 30: 557–563, 2009. 317. Ohno S, Zankov DP, Ding WG, Itoh H, Makiyama T, Doi T, Shizuta S, Hattori T, Miyamoto A, Naiki N, Hancox JC, Matsuura H, Horie M. KCNE5 (KCNE1L) variants are novel modulators of Brugada syndrome and idiopathic ventricular fibrillation. Circ Arrhythm Electrophysiol 4: 352–361, 2011. 318. Okishige K, Ohkubo T, Goseki Y, Matsubara T, Hiejima K, Ibukiyama C. Experimental study of the effects of multi-site sequential ventricular pacing on the prophylaxis of ventricular fibrillation. Jpn Heart J 41: 193–204, 2000. 319. Olesen MS, Bentzen BH, Nielsen JB, Steffensen AB, David JP, Jabbari J, Jensen HK, Haunsø S, Svendsen JH, Schmitt N. Mutations in the potassium channel subunit KCNE1 are associated with early-onset familial atrial fibrillation. BMC Med Genet 13: 24, 2012. 320. Olesen MS, Jabbari J, Holst AG, Nielsen JB, Steinbruchel DA, Jespersen T, Haunso S, Svendsen JH. Screening of KCNN3 in patients with early-onset lone atrial fibrillation. Europace 13: 963–967, 2011. Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 286. Moe GK, Rheinboldt WC, Abildskov JA. A computer model of atrial fibrillation. Am Heart J 67: 200 –220, 1964. 302. Nattel S, Yue L, Wang Z. Cardiac ultrarapid delayed rectifiers: a novel potassium current family o f functional similarity and molecular diversity. Cell Physiol Biochem 9: 217–226, 1999. CARDIAC POTASSIUM CHANNELS AND ARRHYTHMIA 321. Olesen MS, Refsgaard L, Holst AG, Larsen AP, Grubb S, Haunsø S, Svendsen JH, Olesen SP, Schmitt N, Calloe K. A novel KCND3 gain-of-function mutation associated with early-onset of persistent lone atrial fibrillation. Cardiovasc Res 98: 488 – 495, 2013. 322. Olson TM, Alekseev AE, Liu XK, Park S, Zingman LV, Bienengraeber M, Sattiraju S, Ballew JD, Jahangir A, Terzic A. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum Mol Genet 15: 2185–2191, 2006. 323. Ono K, Iijima T. Cardiac T-type Ca2⫹ channels in the heart. J Mol Cell Cardiol 48: 65–70, 2010. 324. Opolski G, Torbicki A, Kosior DA, Szulc M, Wozakowska-Kaplon B, Kolodziej P, Achremczyk P. Rate control vs rhythm control in patients with nonvalvular persistent atrial fibrillation: the results of the Polish How to Treat Chronic Atrial Fibrillation (HOT CAFE) Study. Chest 126: 476 – 486, 2004. 325. Ordög B, Brutyó E, Puskás LG, Papp JG, Varró A, Szabad J, Boldogköi Z. Gene expression profiling of human cardiac potassium and sodium channels. Int J Cardiol 111: 386 –393, 2006. 327. Ozaita A, Vega-Saenz de Miera E. Cloning of two transcripts, HKT4.1a and HKT41b, from the human two-pore K⫹ channel gene KCNK4 Chromosomal localization, tissue distribution and functional expression. Brain Res 102: 18 –27, 2002. 328. Ozgen N, Dun W, Sosunov EA, Anyukhovsky EP, Hirose M, Duffy HS, Boyden PA, Rosen MR. Early electrical remodeling in rabbit pulmonary vein results from trafficking of intracellular SK2 channels to membrane sites. Cardiovasc Res 75: 758 –769, 2007. 329. Pabon A, Chan KW, Sui JL, Wu X, Logothetis DE, Thornhill WB. Glycosylation of GIRK1 at Asn119 and ROMK1 at Asn117 has different consequences in potassium channel function. J Biol Chem 275: 30677–30682, 2000. 330. Pai GR, Rawles JM. The QT interval in atrial fibrillation. Br Heart J 61: 510 –513, 1989. 331. Pandit SV, Berenfeld O, Anumonwo JMB, Zaritski RM, Kneller J, Nattel S, Jalife J. Ionic determinants of functional reentry in a 2-D model of human atrial cells during simulated chronic atrial fibrillation. Biophys J 88: 3806 –3821, 2005. 332. Panguluri SK, Tur J, Chapalamadugu KC, Katnik C, Cuevas J, Tipparaju SM. MicroRNA-301a mediated regulation of Kv4.2 in diabetes: identification of key modulators. PloS One 8: e60545, 2013. 333. Patel AJ, Honoré E. Properties and modulation of mammalian 2P domain K⫹ channels. Trends Neurosci 24: 339 –346, 2001. 334. Patel C, Antzelevitch C. Pharmacological approach to the treatment of long and short QT syndromes. Pharmacol Ther 118: 138 –151, 2008. 335. Patel SP, Campbell DL. Transient outward potassium current, “Ito”, phenotypes in the mammalian left ventricle: underlying molecular, cellular and biophysical mechanisms. J Physiol 569: 7–39, 2005. 336. Pavri BB, Greenberg HE, Kraft WK, Lazarus N, Lynch JJ, Salata JJ, Bilodeau MT, Regan CP, Stump G, Fan L, Mehta A, Wagner JA, Gutstein DE, Bloomfield D. MK0448, a specific Kv1.5 inhibitor: safety, pharmacokinetics, and pharmacodynamic electrophysiology in experimental animal models and humans. Circ Arrhythm Electrophysiol 5: 1193–1201, 2012. 337. Pedersen OD, Bagger H, Keller N, Marchant B, Kober L, Torp-Pedersen C. Efficacy of dofetilide in the treatment of atrial fibrillation-flutter in patients with reduced left ventricular function: a Danish investigations of arrhythmia and mortality on dofetilide (diamond) substudy. Circulation 104: 292–296, 2001. 338. Péréon Y, Demolombe S, Baró I, Drouin E, Charpentier F, Escande D. Differential expression of KvLQT1 isoforms across the human ventricular wall. Am J Physiol Heart Circ Physiol 278: H1908 –H1915, 2000. 339. Perez-Reyes E. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev 83: 117–161, 2003. 343. Pfeufer A, Sanna S, Arking DE, Müller M, Gateva V, Fuchsberger C, Ehret GB, Orrú M, Pattaro C, Köttgen A, Perz S, Usala G, Barbalic M, Li M, Pütz B, Scuteri A, Prineas RJ, Sinner MF, Gieger C, Najjar SS, Kao WHL, Mühleisen TW, Dei M, Happle C, Möhlenkamp S, Crisponi L, Erbel R, Jöckel KH, Naitza S, Steinbeck G, Marroni F, Hicks AA, Lakatta E, Müller-Myhsok B, Pramstaller PP, Wichmann HE, Schlessinger D, Boerwinkle E, Meitinger T, Uda M, Coresh J, Kääb S, Abecasis GR, Chakravarti A. Common variants at ten loci modulate the QT interval duration in the QTSCD Study. Nat Genet 41: 407– 414, 2009. 344. Piccini JP, Hasselblad V, Peterson ED, Washam JB, Califf RM, Kong DF. Comparative efficacy of dronedarone and amiodarone for the maintenance of sinus rhythm in patients with atrial fibrillation. J Am Coll Cardiol 54: 1089 –1095, 2009. 345. Pinto JM, Boyden PA. Reduced inward rectifying and increased E-4031-sensitive K⫹ current density in arrhythmogenic subendocardial purkinje myocytes from the infarcted heart. J Cardiovasc Electrophysiol 9: 299 –311, 1998. 346. Piper DR, Hinz WA, Tallurri CK, Sanguinetti MC, Tristani-Firouzi M. Regional specificity of human ether-a’-go-go-related gene channel activation and inactivation gating. J Biol Chem 280: 7206 –7217, 2005. 347. Pizzale S, Gollob MH, Gow R, Birnie DH. Sudden death in a young man with catecholaminergic polymorphic ventricular tachycardia and paroxysmal atrial fibrillation. J Cardiovasc Electrophysiol 19: 1319 –1321, 2008. 348. Plaster NM, Tawil R, Tristani-Firouzi M, Canún S, Bendahhou S, Tsunoda A, Donaldson MR, Iannaccone ST, Brunt E, Barohn R, Clark J, Deymeer F, George AL Jr, Fish FA, Hahn A, Nitu A, Ozdemir C, Serdaroglu P, Subramony SH, Wolfe G, Fu YH, Ptácek LJ. Mutations in Kir2.1cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell 105: 511–519, 2001. 349. Po SS, Wu RC, Juang GJ, Kong W, Tomaselli GF. Mechanism of alpha-adrenergic regulation of expressed hKv4.3 currents. Am J Physiol Heart Circ Physiol 281: H2518 – H2527, 2001. 350. Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: Roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ Res 88: 1159 – 1167, 2001. 351. Polontchouk L, Haefliger JA, Ebelt B, Schaefer T, Stuhlmann D, Mehlhorn U, KuhnRegnier F, De Vivie ER, Dhein S. Effects of chronic atrial fibrillation on gap junction distribution in human and rat atria. J Am Coll Cardiol 38: 883– 891, 2001. 352. Post W, Shen H, Damcott C, Arking DE, Kao WHL, Sack PA, Ryan KA, Chakravarti A, Mitchell BD, Shuldiner AR. Associations between genetic variants in the NOS1AP (CAPON) gene and cardiac repolarization in the old order Amish. Hum Hered 64: 214 –219, 2007. 353. Pratt CM, Camm AJ, Cooper W, Friedman PL, MacNeil DJ, Moulton KM, Pitt B, Schwartz PJ, Veltri EP, Waldo AL. Mortality in the Survival With ORal D-sotalol (SWORD) trial: why did patients die? Am J Cardiol 81: 869 – 876, 1998. 354. Pribnow D, Johnson-Pais T, Bond CT, Keen J, Johnson RA, Janowsky A, Silvia C, Thayer M, Maylie J, Adelman JP. Skeletal muscle and small-conductance calciumactivated potassium channels. Muscle Nerve 22: 742–750, 1999. 355. Priori SG, Pandit SV, Rivolta I, Berenfeld O, Ronchetti E, Dhamoon A, Napolitano C, Anumonwo J, Barletta di MR, Gudapakkam S, Bosi G, Stramba-Badiale M, Jalife J. A Novel Form of Short QT Syndrome (SQT3) Is Caused by a Mutation in the KCNJ2 Gene. Circ Res 96: 800 – 807, 2005. 356. Putzke C, Wemhöner K, Sachse FB, Rinné S, Schlichthörl G, Li XT, Jaé L, Eckhardt I, Wischmeyer E, Wulf H, Preisig-Müller R, Daut J, Decher N. The acid-sensitive potassium channel TASK-1 in rat cardiac muscle. Cardiovasc Res 75: 59 – 68, 2007. 340. Pertsov AM, Davidenko JM, Salomonsz R, Baxter WT, Jalife J. Spiral waves of excitation underlie reentrant activity in isolated cardiac muscle. Circ Res 72: 631– 650, 1993. 357. Qi XY, Diness JG, Brundel B, Zhou XB, Naud P, Wu CT, Huang H, Harada M, Aflaki M, Dobrev D, Grunnet M, Nattel S. Role of small conductance calcium-activated potassium channels in atrial electrophysiology and fibrillation in the dog. Circulation doi:10.1161/CIRCULATIONAHA.113.003019. 341. Petrecca K, Atanasiu R, Akhavan A, Shrier A. N-linked glycosylation sites determine HERG channel surface membrane expression. J Physiol 515: 41– 48, 1999. 358. Ravens U, Dobrev D. Cardiac sympathetic innervation and control of potassium channel function. J Mol Cell Cardiol 35: 137–139, 2003. Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org 649 Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 326. Osawa M, Kimura R, Hasegawa I, Mukasa N, Satoh F. SNP association and sequence analysis of the NOS1AP gene in SIDS. Leg Med Tokyo Jpn 11 Suppl 1: S307–308, 2009. 342. Petric S, Clasen L, van Wessel C, Geduldig N, Ding Z, Schullenberg M, Mersmann J, Zacharowski K, Aller MI, Schmidt KG, Donner BC. In vivo electrophysiological characterization of TASK-1 deficient mice. Cell Physiol Biochem 30: 523–537, 2012. SCHMITT, GRUNNET, AND OLESEN 359. Ravens U, Wettwer E. Ultra-rapid delayed rectifier channels: molecular basis and therapeutic implications. Cardiovasc Res 89: 776 –785, 2011. 378. Satoh T, Zipes DP. Cesium-induced atrial tachycardia degenerating into atrial fibrillation in dogs: atrial torsades de pointes? J Cardiovasc Electrophysiol 9: 970 –975, 1998. 360. Ravn LS, Aizawa Y, Pollevick GD, Hofman-Bang J, Cordeiro JM, Dixen U, Jensen G, Wu Y, Burashnikov E, Haunso S, Guerchicoff A, Hu D, Svendsen JH, Christiansen M, Antzelevitch C. Gain of function in IKs secondary to a mutation in KCNE5 associated with atrial fibrillation. Heart Rhythm 5: 427– 435, 2008. 379. Schmidt C, Wiedmann F, Schweizer PA, Becker R, Katus HA, Thomas D. Novel electrophysiological properties of dronedarone: inhibition of human cardiac twopore-domain potassium (K2P) channels. Naunyn-Schmiedebergs Arch Pharmacol 385: 1003–1016, 2012. 361. Regan CP, Wallace AA, Cresswell HK, Atkins CL, Lynch JJ Jr. In vivo cardiac electrophysiologic effects of a novel diphenylphosphine oxide IKur blocker, (2-isopropyl-5methylcyclohexyl)diphenylphosphine oxide, in rat and nonhuman primate. J Pharmacol Exp Ther 316: 727–732, 2006. 380. Schram G, Pourrier M, Melnyk P, Nattel S. Differential distribution of cardiac ion channel expression as a basis for regional specialization in electrical function. Circ Res 90: 939 –950, 2002. 362. Riccio ML, Koller ML, Gilmour RF Jr. Electrical restitution and spatiotemporal organization during ventricular fibrillation. Circ Res 84: 955–963, 1999. 363. Risgaard B, Jabbari R, Refsgaard L, Holst AG, Haunsø S, Sadjadieh A, Winkel BG, Olesen MS, Tfelt-Hansen J. High prevalence of genetic variants previously associated with Brugada syndrome in new exome data. Clin Genet 84: 489 – 495, 2013. 381. Schwartz PJ, Priori SG, Napolitano C. How really rare are rare diseases?: the intriguing case of independent compound mutations in the long QT syndrome. J Cardiovasc Electrophysiol 14: 1120 –1121, 2003. 382. Schwetz TA, Norring SA, Bennett ES. N glycans modulate K(v)1.5 gating but have no effect on K(v)1.4 gating. Biochim Biophys Acta 1798: 367–375, 2010. 383. Schwinger RH, Münch G, Bölck B, Karczewski P, Krause EG, Erdmann E. Reduced Ca2⫹-sensitivity of SERCA 2a in failing human myocardium due to reduced serin-16 phospholamban phosphorylation. J Mol Cell Cardiol 31: 479 – 491, 1999. 365. Rogart RB, Cribbs LL, Muglia LK, Kephart DD, Kaiser MW. Molecular cloning of a putative tetrodotoxin-resistant rat heart Na⫹ channel isoform. Proc Natl Acad Sci USA 86: 8170 – 8174, 1989. 384. Seebohm G, Pusch M, Chen J, Sanguinetti MC. Pharmacological activation of normal and arrhythmia-associated mutant KCNQ1 potassium channels. Circ Res 93: 941–947, 2003. 366. Rosa JC, Galanakis D, Ganellin CR, Dunn PM, Jenkinson DH. Bis-quinolinium cyclophanes: 6,10-diaza-3(1,3),8(1,4)-dibenzena-1,5(1,4)- diquinolinacyclodecaphane (UCL 1684), the first nanomolar, non-peptidic blocker of the apamin-sensitive Ca(2⫹)-activated K⫹ channel. J Med Chem 41: 2–5, 1998. 385. Sergeant GP, Ohya S, Reihill JA, Perrino BA, Amberg GC, Imaizumi Y, Horowitz B, Sanders KM, Koh SD. Regulation of Kv4.3currents by Ca2⫹/calmodulin-dependent protein kinase II. Am J Physiol Cell Physiol 288: C304 –C313, 2005. 367. Rosati B, Pan Z, Lypen S, Wang HS, Cohen I, Dixon JE, McKinnon D. Regulation of KChIP2 potassium channel beta subunit gene expression underlies the gradient of transient outward current in canine and human ventricle. J Physiol 533: 119 –125, 2001. 368. Rose J, Armoundas AA, Tian Y, DiSilvestre D, Burysek M, Halperin V, O’Rourke B, Kass DA, Marbán E, Tomaselli GF. Molecular correlates of altered expression of potassium currents in failing rabbit myocardium. Am J Physiol Heart Circ Physiol 288: H2077–H2087, 2005. 369. Rozanski GJ, Xu Z, Whitney RT, Murakami H, Zucker IH. Electrophysiology of rabbit ventricular myocytes following sustained rapid ventricular pacing. J Mol Cell Cardiol 29: 721–732, 1997. 370. Sah R, Ramirez RJ, Oudit GY, Gidrewicz D, Trivieri MG, Zobel C, Backx PH. Regulation of cardiac excitation-contraction coupling by action potential repolarization: role of the transient outward potassium current [I(to)]. J Physiol 546: 5–18, 2003. 371. Salata JJ, Jurkiewicz NK, Wang J, Evans BE, Orme HT, Sanguinetti MC. A novel benzodiazepine that activates cardiac slow delayed rectifier K⫹ currents. Mol Pharmacol 54: 220 –230, 1998. 372. Sale H, Wang J, O’Hara TJ, Tester DJ, Phartiyal P, He JQ, Rudy Y, Ackerman MJ, Robertson GA. Physiological properties of hERG 1a/1b heteromeric currents and a hERG 1b-specific mutation associated with Long-QT syndrome. Circ Res 103: e81–95, 2008. 373. Samie FH, Berenfeld O, Anumonwo J, Mironov SF, Udassi S, Beaumont J, Taffet S, Pertsov AM, Jalife J. Rectification of the background potassium current: a determinant of rotor dynamics in ventricular fibrillation. Circ Res 89: 1216 –1223, 2001. 374. Sande JB, Sjaastad I, Hoen IB, Bøkenes J, Tønnessen T, Holt E, Lunde PK, Christensen G. Reduced level of serine(16) phosphorylated phospholamban in the failing rat myocardium: a major contributor to reduced SERCA2 activity. Cardiovasc Res 53: 382– 391, 2002. 375. Sanders P, Kistler PM, Morton JB, Spence SJ, Kalman JM. Remodeling of sinus node function in patients with congestive heart failure: reduction in sinus node reserve. Circulation 110: 897–903, 2004. 376. Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 384: 80 – 83, 1996. 377. Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature 440: 463– 469, 2006. 650 386. Seyler C, Duthil-Straub E, Zitron E, Gierten J, Scholz EP, Fink RHA, Karle CA, Becker R, Katus HA, Thomas DT. ASK1 (K(2P)3.1) K(⫹) channel inhibition by endothelin-1 is mediated through Rho kinase-dependent phosphorylation. Br J Pharmacol 165: 1467– 1475, 2012. 387. Shan H, Zhang Y, Cai B, Chen X, Fan Y, Yang L, Chen X, Liang H, Zhang Y, Song X, Xu C, Lu Y, Yang B, Du Z. Upregulation of microRNA-1 and microRNA-133 contributes to arsenic-induced cardiac electrical remodeling. Int J Cardiol 167: 2798 –2805, 2013. 388. Shannon TR, Pogwizd SM, Bers DM. Elevated sarcoplasmic reticulum Ca2⫹ leak in intact ventricular myocytes from rabbits in heart failure. Circ Res 93: 592–594, 2003. 389. Shantsila E, Watson T, Lip GYH. Drug-induced QT-interval prolongation and proarrhythmic risk in the treatment of atrial arrhythmias. Europace 9 Suppl 4: iv37– 44, 2007. 390. Shi Y, Ducharme A, Li D, Gaspo R, Nattel S, Tardif JC. Remodeling of atrial dimensions and emptying function in canine models of atrial fibrillation. Cardiovasc Res 52: 217– 225, 2001. 391. Shinagawa K, Shi YF, Tardif JC, Leung TK, Nattel S. Dynamic nature of atrial fibrillation substrate during development and reversal of heart failure in dogs. Circulation 105: 2672–2678, 2002. 392. Shorofsky SR, January CT. L- and T-type Ca2⫹ channels in canine cardiac Purkinje cells. Single-channel demonstration of L-type Ca2⫹ window current. Circ Res 70: 456 – 464, 1992. 393. Sills MN, Xu YC, Baracchini E, Goodman RH, Cooperman SS, Mandel G, Chien KR. Expression of diverse Na⫹ channel messenger RNAs in rat myocardium. Evidence for a cardiac-specific Na⫹ channel. J Clin Invest 84: 331–336, 1989. 394. Sinner MF, Ellinor PT, Meitinger T, Benjamin EJ, Kääb S. Genome-wide association studies of atrial fibrillation: past, present, and future. Cardiovasc Res 89: 701–709, 2011. 395. Skanes AC, Gray RA, Zuur CL, Jalife J. Effects of postshock atrial pacing on atrial defibrillation outcome in the isolated sheep heart. Circulation 98: 64 –72, 1998. 396. Skibsbye L, Diness JG, Sørensen US, Hansen RS, Grunnet M. The duration of pacinginduced atrial fibrillation is reduced in vivo by inhibition of small conductance Ca2⫹activated K⫹ channels. J Cardiovasc Pharmacol 57: 672– 681, 2011. 397. Snyders DJ, Tamkun MM, Bennett PB. A rapidly activating and slowly inactivating potassium channel cloned from human heart. Functional analysis after stable mammalian cell culture expression. J Gen Physiol 101: 513–543, 1993. Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 364. Roden DM. Taking the “idio” out of “idiosyncratic”: predicting torsades de pointes. Pacing Clin Electrophysiol 21: 1029 –1034, 1998. CARDIAC POTASSIUM CHANNELS AND ARRHYTHMIA 398. Soldatov NM. Genomic structure of human L-type Ca2⫹ channel. Genomics 22: 77– 87, 1994. 399. Soltysinska E, Olesen SP, Christ T, Wettwer E, Varró A, Grunnet M, Jespersen T. Transmural expression of ion channels and transporters in human nondiseased and end-stage failing hearts. Pflügers Arch 459: 11–23, 2009. 400. Sorensen US, Strobaek D, Christophersen P, Hougaard C, Jensen ML, Nielsen EO, Peters D, Teuber L. Synthesis and structure-activity relationship studies of 2-(Nsubstituted)-aminobenzimidazoles as potent negative gating modulators of small conductance Ca2⫹-activated K⫹ channels. J Med Chem 51: 7625–7634, 2008. 417. Tanaka H, Miake J, Notsu T, Sonyama K, Sasaki N, Iitsuka K, Kato M, Taniguchi S, Igawa O, Yoshida A, Shigemasa C, Hoshikawa Y, Kurata Y, Kuniyasu A, Nakayama H, Inagaki N, Nanba E, Shiota G, Morisaki T, Ninomiya H, Kitakaze M, Hisatome I. Proteasomal degradation of Kir6.2 channel protein and its inhibition by a Na⫹ channel blocker aprindine. Biochem Biophys Res Commun 331: 1001–1006, 2005. 418. Tanaka H, Namekata I, Hamaguchi S, Kawamura T, Masuda H, Tanaka Y, Iida-Tanaka N, Takahara A. Effect of NIP-142 on potassium channel alpha-subunits Kv1.5, Kv42 and Kv43, and mouse atrial repolarization. Biol Pharm Bull 33: 138 –141, 2010. 419. Thomas D, Zhang W, Karle CA, Kathöfer S, Schöls W, Kübler W, Kiehn J. Deletion of protein kinase A phosphorylation sites in the HERG potassium channel inhibits activation shift by protein kinase A. J Biol Chem 274: 27457–27462, 1999. 420. Thusberg J, Olatubosun A, Vihinen M. Performance of mutation pathogenicity prediction methods on missense variants. Hum Mutat 32: 358 –368, 2011. 402. Splawski I, Shen J, Timothy KW, Vincent GM, Lehmann MH, Keating MT. Genomic structure of three long QT syndrome genes: KVLQT1, HERG, and KCNE1. Genomics 51: 86 –97, 1998. 421. Töpert C, Döring F, Wischmeyer E, Karschin C, Brockhaus J, Ballanyi K, Derst C, Karschin A. Kir2.4:a novel K⫹ inward rectifier channel associated with motoneurons of cranial nerve nuclei. J Neurosci 18: 4096 – 4105, 1998. 403. Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat Genet 17: 338 –340, 1997. 422. Trudeau MC, Leung LM, Roti ER, Robertson GA. hERG1a N-terminal eag domaincontaining polypeptides regulate homomeric hERG1b and heteromeric hERG1a/ hERG1b channels: a possible mechanism for long QT syndrome. J Gen Physiol 138: 581–592, 2011. 404. Staudacher K, Baldea I, Kisselbach J, Staudacher I, Rahm AK, Schweizer PA, Becker R, Katus HA, Thomas D. Alternative splicing determines mRNA translation initiation and function of human K(2P)10.1 K⫹ channels. J Physiol 589: 3709 –3720, 2011. 405. Staudacher K, Staudacher I, Ficker E, Seyler C, Gierten J, Kisselbach J, Rahm AK, Trappe K, Schweizer PA, Becker R, Katus HA, Thomas D. Carvedilol targets human K2P 3.1 (TASK1) K⫹ leak channels. Br J Pharmacol 163: 1099 –1110, 2011. 423. Tsuji Y, Opthof T, Kamiya K, Yasui K, Liu W, Lu Z, Kodama I. Pacing-induced heart failure causes a reduction of delayed rectifier potassium currents along with decreases in calcium and transient outward currents in rabbit ventricle. Cardiovasc Res 48: 300 – 309, 2000. 406. Stocker M. Ca -activated K channels: molecular determinants and function of the SK family. Nat Rev Neurosci 5: 758 –770, 2004. 424. Tsuji Y, Zicha S, Qi XY, Kodama I, Nattel S. Potassium channel subunit remodeling in rabbits exposed to long-term bradycardia or tachycardia: discrete arrhythmogenic consequences related to differential delayed-rectifier changes. Circulation 113: 345– 355, 2006. 407. Streit AK, Netter MF, Kempf F, Walecki M, Rinné S, Bollepalli MK, Preisig-Müller R, Renigunta V, Daut J, Baukrowitz T, Sansom MSP, Stansfeld PJ, Decher N. A specific two-pore domain potassium channel blocker defines the structure of the TASK-1 open pore. J Biol Chem 286: 13977–13984, 2011. 425. Tuteja D, Rafizadeh S, Timofeyev V, Wang S, Zhang Z, Li N, Mateo RK, Singapuri A, Young JN, Knowlton AA, Chiamvimonvat N. Cardiac small conductance Ca2⫹-activated K⫹ channel subunits form heteromultimers via the coiled-coil domains in the C termini of the channels. Circ Res 107: 851– 859, 2010. 408. Strøbaek D, Hougaard C, Johansen TH, Sørensen US, Nielsen EØ, Nielsen KS, Taylor RDT, Pedarzani P, Christophersen P. Inhibitory gating modulation of small conductance Ca2⫹-activated K⫹ channels by the synthetic compound (R)-N-(benzimidazol2-yl)-1,2,3,4-tetrahydro-1-naphtylamine (NS8593) reduces afterhyperpolarizing current in hippocampal CA1 neurons. Mol Pharmacol 70: 1771–1782, 2006. 426. Tuteja D, Xu D, Timofeyev V, Lu L, Sharma D, Zhang Z, Xu Y, Nie L, Vazquez AE, Young JN, Glatter KA, Chiamvimonvat N. Differential expression of small-conductance Ca2⫹-activated K⫹ channels SK1, SK2, and SK3 in mouse atrial and ventricular myocytes. Am J Physiol Heart Circ Physiol 289: H2714 –H2723, 2005. 2⫹ ⫹ 409. Strobaek D, Jorgensen TD, Christophersen P, Ahring PK, Olesen SP. Pharmacological characterization of small-conductance Ca2⫹-activated K⫹ channels stably expressed in HEK 293 cells. Br J Pharmacol 129: 991–999, 2000. 410. Stump MR, Gong Q, Packer JD, Zhou Z. Early LQT2 nonsense mutation generates N-terminally truncated hERG channels with altered gating properties by the reinitiation of translation. J Mol Cell Cardiol 53: 725–733, 2012. 411. Su Z, Limberis J, Souers A, Kym P, Mikhail A, Houseman K, Diaz G, Liu X, Martin RL, Cox BF, Gintant GA. Electrophysiologic characterization of a novel hERG channel activator. Biochem Pharmacol 77: 1383–1390, 2009. 412. Swissa M, Qu Z, Ohara T, Lee MH, Lin SF, Garfinkel A, Karagueuzian HS, Weiss JN, Chen PS. Action potential duration restitution and ventricular fibrillation due to rapid focal excitation. Am J Physiol Heart Circ Physiol 282: H1915–H1923, 2002. 413. Takahashi N, Morishige K, Jahangir A, Yamada M, Findlay I, Koyama H, Kurachi Y. Molecular cloning and functional expression of cDNA encoding a second class of inward rectifier potassium channels in the mouse brain. J Biol Chem 269: 23274 – 23279, 1994. 414. Takigawa M, Kawamura M, Noda T, Yamada Y, Miyamoto K, Okamura H, Satomi K, Aiba T, Kamakura S, Sakaguchi T, Mizusawa Y, Itoh H, Horie M, Shimizu W. Seasonal and circadian distributions of cardiac events in genotyped patients with congenital long QT syndrome. Circ J 76: 2112–2118, 2012. 427. Ueda N, Zipes DP, Wu J. Epicardial but not endocardial premature stimulation initiates ventricular tachyarrhythmia in canine in vitro model of long QT syndrome. Heart Rhythm 1: 684 – 694, 2004. 428. Ufret-Vincenty CA, Baro DJ, Lederer WJ, Rockman HA, Quinones LE, Santana LF. Role of sodium channel deglycosylation in the genesis of cardiac arrhythmias in heart failure. J Biol Chem 276: 28197–28203, 2001. 429. Umigai N, Sato Y, Mizutani A, Utsumi T, Sakaguchi M, Uozumi N. Topogenesis of two transmembrane type K⫹ channels, Kir 2.1 and KcsA. J Biol Chem 278: 40373– 40384, 2003. 430. Undrovinas AI, Maltsev VA, Sabbah HN. Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: role of sustained inward current. Cell Mol Life Sci 55: 494 –505, 1999. 431. Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, Kamp TJ, Makielski JC. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol 38: 475– 483, 2005. 432. Vandenberg CA. Inward rectification of a potassium channel in cardiac ventricular cells depends on internal magnesium ions. Proc Natl Acad Sci USA 84: 2560 –2564, 1987. 433. Vandenberg JI, Perry MD, Perrin MJ, Mann SA, Ke Y, Hill AP. hERG K⫹ channels: structure, function, and clinical significance. Physiol Rev 92: 1393–1478, 2012. 415. Tamkun MM, Knoth KM, Walbridge JA, Kroemer H, Roden DM, Glover DM. Molecular cloning and characterization of two voltage-gated K⫹ channel cDNAs from human ventricle. FASEB J 5: 331–337, 1991. 434. Venetucci LA, Trafford AW, O’Neill SC, Eisner DA. Na/Ca exchange: regulator of intracellular calcium and source of arrhythmias in the heart. Ann NY Acad Sci 1099: 315–325, 2007. 416. Tanaka H, Hashimoto N. A multiple ion channel blocker, NIP-142, for the treatment of atrial fibrillation. Cardiovasc Rev 25: 342–356, 2007. 435. Vergara C, Latorre R, Marrion NV, Adelman JP. Calcium-activated potassium channels. Curr Opin Neurobiol 8: 321–329, 1998. Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org 651 Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 401. Sossalla S, Kallmeyer B, Wagner S, Mazur M, Maurer U, Toischer K, Schmitto JD, Seipelt R, Schöndube FA, Hasenfuss G, Belardinelli L, Maier LS. Altered Na⫹ currents in atrial fibrillation effects of ranolazine on arrhythmias and contractility in human atrial myocardium. J Am Coll Cardiol 55: 2330 –2342, 2010. SCHMITT, GRUNNET, AND OLESEN 436. Verkerk AO, Remme CA, Schumacher CA, Scicluna BP, Wolswinkel R, de Jonge B, Bezzina CR, Veldkamp MW. Functional Nav1.8 channels in intracardiac neurons: the link between SCN10A and cardiac electrophysiology. Circ Res 111: 333–343, 2012. 455. Wijffels MC, Dorland R, Mast F, Allessie MA. Widening of the excitable gap during pharmacological cardioversion of atrial fibrillation in the goat: effects of cibenzoline, hydroquinidine, flecainide, and D-sotalol. Circulation 102: 260 –267, 2000. 437. Vick JA, Hassett CC, Shipman WH. Beta adrenergic and antiarrhythmic effect of apamin, a component of bee venom. Edgewood Arsenal Tech Rep 4664: 1–13, 1972. 456. Wijffels MC, Kirchhof CJ, Dorland R, Allessie MA. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation 92: 1954 –1968, 1995. 438. Voigt N, Maguy A, Yeh YH, Qi X, Ravens U, Dobrev D, Nattel S. Changes in IK,ACh single-channel activity with atrial tachycardia remodelling in canine atrial cardiomyocytes. Cardiovasc Res 77: 35– 43, 2008. 439. Van Wagoner DR, Pond AL, McCarthy PM, Trimmer JS, Nerbonne JM. Outward K⫹ current densities and Kv1.5 expression are reduced in chronic human atrial fibrillation. Circ Res 80: 772–781, 1997. 440. Waldo AL, Camm AJ, deRuyter H, Friedman PL, MacNeil DJ, Pauls JF, Pitt B, Pratt CM, Schwartz PJ, Veltri EP. Effect of D-sotalol on mortality in patients with left ventricular dysfunction after recent and remote myocardial infarction. The SWORD Investigators Survival With Oral D-Sotalol. Lancet 348: 7–12, 1996. 442. Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, de JT, Schwartz PJ, Toubin JA, Moss AJ, Atkinson DL, Landes GM, Connors TD, Keating MT. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet 12: 17–23, 1996. 458. Winfree AT. Varieties of spiral wave behavior: an experimentalist’s approach to the theory of excitable media. Chaos Woodbury N 1: 303–334, 1991. 459. Wischmeyer E, Karschin A. Receptor stimulation causes slow inhibition of IRK1 inwardly rectifying K⫹ channels by direct protein kinase A-mediated phosphorylation. Proc Natl Acad Sci USA 93: 5819 –5823, 1996. 460. Wittekindt OH, Visan V, Tomita H, Imtiaz F, Gargus JJ, Lehmann-Horn F, Grissmer S, Morris-Rosendahl DJ. An apamin- and scyllatoxin-insensitive isoform of the human SK3 channel. Mol Pharmacol 65: 788 – 801, 2004. 461. Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke 22: 983–988, 1991. 462. Workman AJ, Kane KA, Rankin AC. The contribution of ionic currents to changes in refractoriness of human atrial myocytes associated with chronic atrial fibrillation. Cardiovasc Res 52: 226 –235, 2001. 443. Wang TJ, Larson MG, Levy D, Vasan RS, Leip EP, Wolf PA, D’Agostino RB, Murabito JM, Kannel WB, Benjamin EJ. Temporal relations of atrial fibrillation and congestive heart failure and their joint influence on mortality: the Framingham Heart Study. Circulation 107: 2920 –2925, 2003. 463. Workman AJ, Marshall GE, Rankin AC, Smith GL, Dempster J. Transient outward K⫹ current reduction prolongs action potentials and promotes afterdepolarisations: a dynamic-clamp study in human and rabbit cardiac atrial myocytes. J Physiol 590: 4289 – 4305, 2012. 444. Wang W, Watanabe M, Nakamura T, Kudo Y, Ochi R. Properties and expression of Ca2⫹-activated K⫹ channels in H9c2 cells derived from rat ventricle. Am J Physiol Heart Circ Physiol 276: H1559 –H1566, 1999. 464. Wrobel E, Tapken D, Seebohm G. The KCNE Tango: how KCNE1 interacts with Kv7.1. Front Pharmacol 3: 142, 2012. 445. Wang X, Liang B, Skibsbye L, Olesen SP, Grunnet M, Jespersen T. GIRK channel activation via adenosine or muscarinic receptors has similar effects on rat atrial electrophysiology. J Cardiovasc Pharmacol 62: 192–198, 2013. 446. Warren M, Guha PK, Berenfeld O, Zaitsev A, Anumonwo JMB, Dhamoon AS, Bagwe S, Taffet SM, Jalife J. Blockade of the inward rectifying potassium current terminates ventricular fibrillation in the guinea pig heart. J Cardiovasc Electrophysiol 14: 621– 631, 2003. 447. Watanabe I, Kanda A, Engle CL, Gettes LS. Comparison of the effects of regional ischemia and hyperkalemia on the membrane action potentials of the in situ pig heart. Experimental Cardiology Group, University of North Carolina at Chapel Hill. J Cardiovasc Electrophysiol 8: 1229 –1236, 1997. 448. Watanabe Y, Hara Y, Tamagawa M, Nakaya H. Inhibitory effect of amiodarone on the muscarinic acetylcholine receptor-operated potassium current in guinea pig atrial cells. J Pharmacol Exp Ther 279: 617– 624, 1996. 449. Weatherall KL, Goodchild SJ, Jane DE, Marrion NV. Small conductance calciumactivated potassium channels: from structure to function. Prog Neurobiol 91: 242–255, 2010. 450. Wei AD, Gutman GA, Aldrich R, Chandy KG, Grissmer S, Wulff H. International Union of Pharmacology. LII. Nomenclature and molecular relationships of calcium-activated potassium channels. Pharmacol Rev 57: 463– 472, 2005. 465. Wulff H, Kolski-Andreaco A, Sankaranarayanan A, Sabatier JM, Shakkottai V. Modulators of small- and intermediate-conductance calcium-activated potassium channels and their therapeutic indications. Curr Med Chem 14: 1437–1457, 2007. 466. Wyse DG, Waldo AL, DiMarco JP, Domanski MJ, Rosenberg Y, Schron EB, Kellen JC, Greene HL, Mickel MC, Dalquist JE, Corley SD. A comparison of rate control and rhythm control in patients with atrial fibrillation. N Engl J Med 347: 1825–1833, 2002. 467. Xia M, Jin Q, Bendahhou S, He Y, Larroque MM, Chen Y, Zhou Q, Yang Y, Liu Y, Liu B, Zhu Q, Zhou Y, Lin J, Liang B, Li L, Dong X, Pan Z, Wang R, Wan H, Qiu W, Xu W, Eurlings P, Barhanin J, Chen YA. Kir 2.1 gain-of-function mutation underlies familial atrial fibrillation. Biochem Biophys Res Commun 332: 1012–1019, 2005. 468. Xia XM, Fakler B, Rivard A, Wayman G, Johnson-Pais T, Keen JE, Ishii T, Hirschberg B, Bond CT, Lutsenko S, Maylie J, Adelman JP. Mechanism of calcium gating in smallconductance calcium-activated potassium channels. Nature 395: 503–507, 1998. 469. Xie C, Bondarenko VE, Morales MJ, Strauss HC. Closed-state inactivation in Kv4.3 isoforms is differentially modulated by protein kinase C. Am J Physiol Cell Physiol 297: C1236 –C1248, 2009. 470. Xu H, Guo W, Nerbonne JM. Four kinetically distinct depolarization-activated K⫹ currents in adult mouse ventricular myocytes. J Gen Physiol 113: 661– 678, 1999. 451. Weidmann S. Effect of current flow on the membrane potential of cardiac muscle. J Physiol 115: 227–236, 1951. 471. Xu Y, Tuteja D, Zhang Z, Xu D, Zhang Y, Rodriguez J, Nie L, Tuxson HR, Young JN, Glatter KA, Vazquez AE, Yamoah EN, Chiamvimonvat N. Molecular identification and functional roles of a Ca2⫹-activated K⫹ channel in human and mouse hearts. J Biol Chem 278: 49085– 49094, 2003. 452. Westenbroek RE, Bischoff S, Fu Y, Maier SKG, Catterall WA, Scheuer T. Localization of sodium channel subtypes in mouse ventricular myocytes using quantitative immunocytochemistry. J Mol Cell Cardiol 64: 69 –78, 2013. 472. Yagi T, Pu J, Chandra P, Hara M, Danilo P Jr, Rosen MR, Boyden PA. Density and function of inward currents in right atrial cells from chronically fibrillating canine atria. Cardiovasc Res 54: 405– 415, 2002. 453. Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Compound mutations: a common cause of severe long-QT syndrome. Circulation 109: 1834 – 1841, 2004. 473. Yamashita T, Murakawa Y, Ajiki K, Omata M. Incidence of induced atrial fibrillation/ flutter in complete atrioventricular block. A concept of “atrial-malfunctioning” atriohisian block. Circulation 95: 650 – 654, 1997. 454. Wettwer E, Hala O, Christ T, Heubach JF, Dobrev D, Knaut M, Varro A, Ravens U. Role of IKur in controlling action potential shape and contractility in the human atrium: influence of chronic atrial fibrillation. Circulation 110: 2299 –2306, 2004. 474. Yamawake N, Hirano Y, Sawanobori T, Hiraoka M. Arrhythmogenic effects of isoproterenol-activated Cl⫺ current in guinea-pig ventricular myocytes. J Mol Cell Cardiol 24: 1047–1058, 1992. 652 Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 441. Wang DW, Yazawa K, George AL Jr, Bennett PB. Characterization of human cardiac Na⫹ channel mutations in the congenital long QT syndrome. Proc Natl Acad Sci USA 93: 13200 –13205, 1996. 457. Winfree A. When Time Breaks Down: The Three-Dimensional Dynamics of Electrochemical Waves and Cardiac Arrhythmias. Princeton, NJ: Princeton Univ. Press, 1987. CARDIAC POTASSIUM CHANNELS AND ARRHYTHMIA 475. Yang T, Atack TC, Stroud DM, Zhang W, Hall L, Roden DM. Blocking Scn10a channels in heart reduces late sodium current and is antiarrhythmic. Circ Res 111: 322–332, 2012. 476. Yang Y, Li J, Lin X, Yang Y, Hong K, Wang L, Liu J, Li L, Yan D, Liang D, Xiao J, Jin H, Wu J, Zhang Y, Chen YH. Novel KCNA5 loss-of-function mutations responsible for atrial fibrillation. J Hum Genet 54: 277–283, 2009. 477. Yang Y, Xia M, Jin Q, Bendahhou S, Shi J, Chen Y, Liang B, Lin J, Liu Y, Liu B, Zhou Q, Zhang D, Wang R, Ma N, Su X, Niu K, Pei Y, Xu W, Chen Z, Wan H, Cui J, Barhanin J, Chen Y. Identification of a KCNE2 gain-of-function mutation in patients with familial atrial fibrillation. Am J Hum Genet 75: 899 –905, 2004. 478. Yang Y, Yang Y, Liang B, Liu J, Li J, Grunnet M, Olesen SP, Rasmussen HB, Ellinor PT, Gao L, Lin X, Li L, Wang L, Xiao J, Liu Y, Liu Y, Zhang S, Liang D, Peng L, Jespersen T, Chen YH. Identification of a Kir3.4 mutation in congenital long QT syndrome. Am J Hum Genet 86: 872– 880, 2010. 489. Zhang DF, Liang B, Lin J, Liu B, Zhou QS, Yang YQ. KCNE3 R53H substitution in familial atrial fibrillation. Chin Med J 118: 1735–1738, 2005. 490. Zhang DY, Wang Y, Lau CP, Tse HF, Li GR. Both EGFR kinase and Src-related tyrosine kinases regulate human ether-à-go-go-related gene potassium channels. Cell Signal 20: 1815–1821, 2008. 491. Zhang DY, Wu W, Deng XL, Lau CP, Li GR. Genistein and tyrphostin AG556 inhibit inwardly-rectifying Kir2.1 channels expressed in HEK 293 cells via protein tyrosine kinase inhibition. Biochim Biophys Acta 1808: 1993–1999, 2011. 492. Zhang H, Garratt CJ, Zhu J, Holden AV. Role of up-regulation of IK1 in action potential shortening associated with atrial fibrillation in humans. Cardiovasc Res 66: 493–502, 2005. 493. Zhang L, Foster K, Li Q, Martens JR. S-acylation regulates Kv1.5 channel surface expression. Am J Physiol Cell Physiol 293: C152–C161, 2007. 494. Zhang Y, Post WS, Blasco-Colmenares E, Dalal D, Tomaselli GF, Guallar E. Electrocardiographic QT interval and mortality: a meta-analysis. Epidemiol Camb Mass 22: 660 – 670, 2011. 480. Yeh YH, Qi X, Shiroshita-Takeshita A, Liu J, Maguy A, Chartier D, Hebert T, Wang Z, Nattel S. Atrial tachycardia induces remodelling of muscarinic receptors and their coupled potassium currents in canine left atrial and pulmonary vein cardiomyocytes. Br J Pharmacol 152: 1021–1032, 2007. 495. Zhang YH, Wu W, Sun HY, Deng XL, Cheng LC, Li X, Tse HF, Lau CP, Li GR. Modulation of human cardiac transient outward potassium current by EGFR tyrosine kinase and Src-family kinases. Cardiovasc Res 93: 424 – 433, 2012. 481. Yeh YH, Wakili R, Qi XY, Chartier D, Boknik P, Kääb S, Ravens U, Coutu P, Dobrev D, Nattel S. Calcium-handling abnormalities underlying atrial arrhythmogenesis and contractile dysfunction in dogs with congestive heart failure. Circ Arrhythm Electrophysiol 1: 93–102, 2008. 482. Yu FH, Catterall WA. Overview of the voltage-gated sodium channel family. Genome Biol 4: 207, 2003. 483. Yu FH, Yarov-Yarovoy V, Gutman GA, Catterall WA. Overview of molecular relationships in the voltage-gated ion channel superfamily. Pharmacol Rev 57: 387–395, 2005. 484. Yu T, Wu RB, Guo HM, Deng CY, Zheng SY, Kuang SJ. Expression and functional role of small conductance Ca2⫹-activated K⫹ channels in human atrial myocytes. Nan Fang Yi Ke Xue Xue Bao 31: 490 – 494, 2011. 496. Zhang Z, Xu Y, Song H, Rodriguez J, Tuteja D, Namkung Y, Shin HS, Chiamvimonvat N. Functional Roles of Ca(v)1.3 [alpha(1D)] calcium channel in sinoatrial nodes: insight gained using gene-targeted null mutant mice. Circ Res 90: 981–987, 2002. 497. Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, Tsuchihashi T, McManus MT, Schwartz RJ, Srivastava D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1–2. Cell 129: 303–317, 2007. 498. Zhou J, ugelli-Szafran CE, Bradley JA, Chen X, Koci BJ, Volberg WA, Sun Z, Cordes JS. Novel potent human ether-a-go-go-related gene (hERG) potassium channel enhancers and their in vitro antiarrhythmic activity. Mol Pharmacol 68: 876 – 884, 2005. 499. Zhu L, Wu X, Wu MB, Chan KW, Logothetis DE, Thornhill WB. Cloning and characterization of G protein-gated inward rectifier K⫹ channel (GIRK1) isoforms from heart and brain. J Mol Neurosci 16: 21–32, 2001. 485. Yue L, Feng J, Gaspo R, Li GR, Wang Z, Nattel S. Ionic remodeling underlying action potential changes in a canine model of atrial fibrillation. Circ Res 81: 512–525, 1997. 500. Zicha S, Fernández-Velasco M, Lonardo G, L’Heureux N, Nattel S. Sinus node dysfunction and hyperpolarization-activated (HCN) channel subunit remodeling in a canine heart failure model. Cardiovasc Res 66: 472– 481, 2005. 486. Zareba W, Moss AJ, Locati EH, Lehmann MH, Peterson DR, Hall WJ, Schwartz PJ, Vincent GM, Priori SG, Benhorin J, Towbin JA, Robinson JL, Andrews ML, Napolitano C, Timothy K, Zhang L, Medina A. Modulating effects of age and gender on the clinical course of long QT syndrome by genotype. J Am Coll Cardiol 42: 103–109, 2003. 501. Zicha S, Xiao L, Stafford S, Cha TJ, Han W, Varro A, Nattel S. Transmural expression of transient outward potassium current subunits in normal and failing canine and human hearts. J Physiol 561: 735–748, 2004. 487. Zarraga IG, Zhang L, Stump MR, Gong Q, Vincent GM, Zhou Z. Nonsense-mediated mRNA decay caused by a frameshift mutation in a large kindred of type 2 long QT syndrome. Heart Rhythm 8: 1200 –1206, 2011. 488. Zeng J, Rudy Y. Early afterdepolarizations in cardiac myocytes: mechanism and rate dependence. Biophys J 68: 949 –964, 1995. 502. Zimmer T, Bollensdorff C, Haufe V, Birch-Hirschfeld E, Benndorf K. Mouse heart Na⫹ channels: primary structure and function of two isoforms and alternatively spliced variants. Am J Physiol Heart Circ Physiol 282: H1007–H1017, 2002. 503. Zygmunt AC, Goodrow RJ, Weigel CM. INaCa and ICl(Ca) contribute to isoproterenolinduced delayed after depolarizations in midmyocardial cells. Am J Physiol Heart Circ Physiol 275: H1979 –H1992, 1998. Physiol Rev • VOL 94 • APRIL 2014 • www.prv.org 653 Downloaded from http://physrev.physiology.org/ by 10.220.33.2 on May 2, 2017 479. Yano M, Yamamoto T, Ikemoto N, Matsuzaki M. Abnormal ryanodine receptor function in heart failure. Pharmacol Ther 107: 377–391, 2005.